Translating chemical intuition to machine learning algorithms: identifying promising next-generation methane storage materials
Translating chemical intuition to machine learning algorithms: identifying promising next-generation methane storage materials
Promotor(en): V. Van Speybroeck, S.M.J. Rogge /25666 / Nanoporous materialsBackground and problem
Nanoporous materials have recently attracted a lot of attention due to their wide range of possible applications, such as gas adsorption and separation, catalysis, and photoelectronic applications. Among these materials are the covalent organic frameworks (COFs) [1], a rather new class of materials, that have already shown much promise in a variety of applications. They are synthesized by organic molecules that are linked by strong covalent bonds, resulting in light-weight materials with an extraordinary stability, which makes them of the utmost interest for industrial applications. One of these applications is the storage of methane gas. When stored in empty vessels, methane has only a limited density, making its storage and transport very inefficient. When first filling the vessels with nanoporous materials such as COFs, however, the favorable interactions between methane and the COF material ensure that the methane molecules can be adsorbed in the pores of the material under a higher density than in the empty vessel. As a result, these COF-filled vessels can be adopted in fuel cells or can be used to capture greenhouse gasses. Within each COF, three types of domains can be identified (see Fig. 1): the cores of the building blocks, the chemical linkages that link these cores and the functional groups that are added to the cores. Together with the selection of an appropriate topology, which describes how the building blocks are connected in a framework structure, a careful choice for each of these domains can tune the performance of the resulting COF.
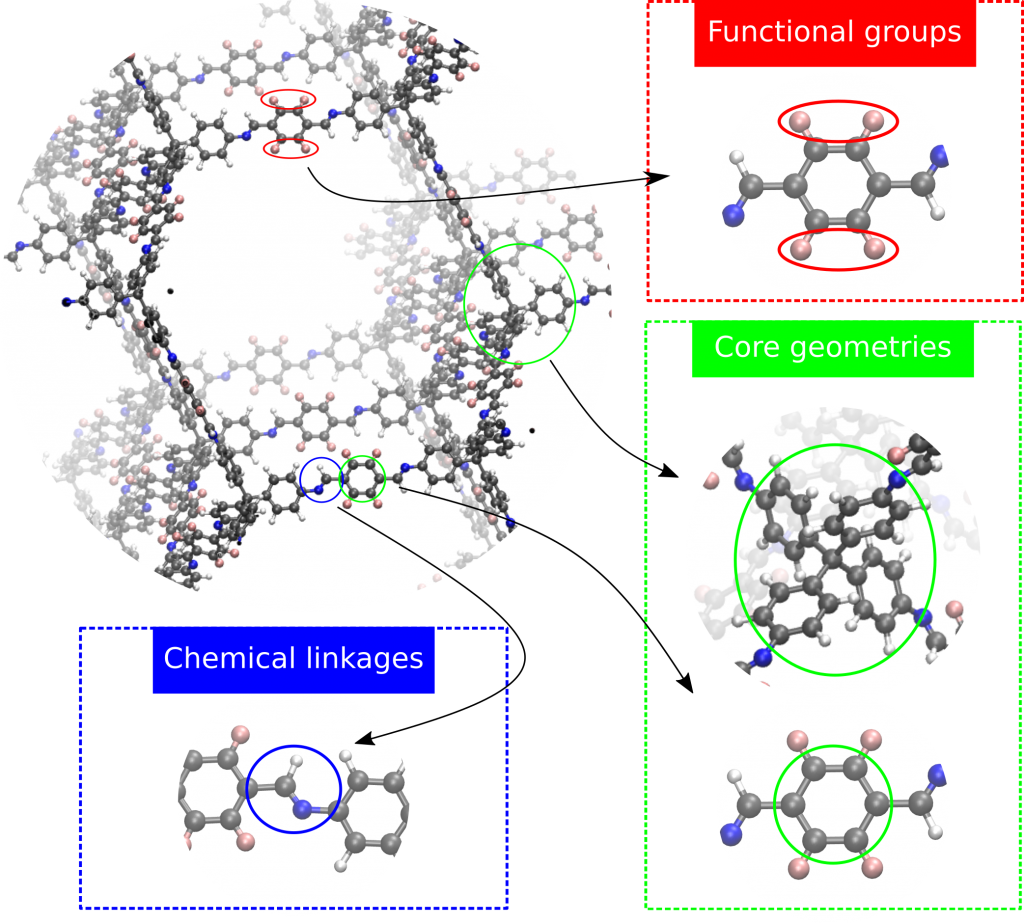
As a result of this building block nature [2] and the large number of each of these domain components that have already been introduced in literature, the number of COFs that can possibly be synthesized is almost unlimited, making an experimental screening of this material class unfeasible. Computational high-throughput screenings [3] offer a valuable alternative as they can characterize a material and identify the most promising materials much faster, which can subsequently be experimentally synthesized. As starting point for these screening studies, a number of COF databases have been constructed, with both experimental and hypothetical COFs [4]. The inclusion of hypothetical structures is important to ensure that the database represents the large versatility observed in COF structures, as experimental databases can be biased towards certain COF subclasses, due to experimental considerations [5]. Recently, a database containing more than 450.000 COFs has been developed in our group, without even considering the variety of functional groups that can be introduced. An extensive screening of a database of this magnitude becomes a cumbersome task, even with the appropriate computational tools.
A solution for this problem is to screen the database using machine learning algorithms (see Fig. 2) [6], where a model is trained on a subset of the database and tries to predict the expected methane uptake from the remaining structures. Special attention has to be paid to the representation of each of the materials as different feature levels can be proposed. When synthesizing new materials for a specific application experimentalists intuitively use knowledge about the individual building blocks to obtain the desired material characteristics. However, establishing reliable information on the effect of certain building blocks in a material requires several successfully synthesized materials which can be critically compared. Therefore, to construct such a knowledge database in silico, the specific goal of this thesis is to propose features that are based solely on the individual components from which the structure is built. This can include the density and pore geometry of the topology, and the molecular structure of the cores, linkages, and functional groups [5].
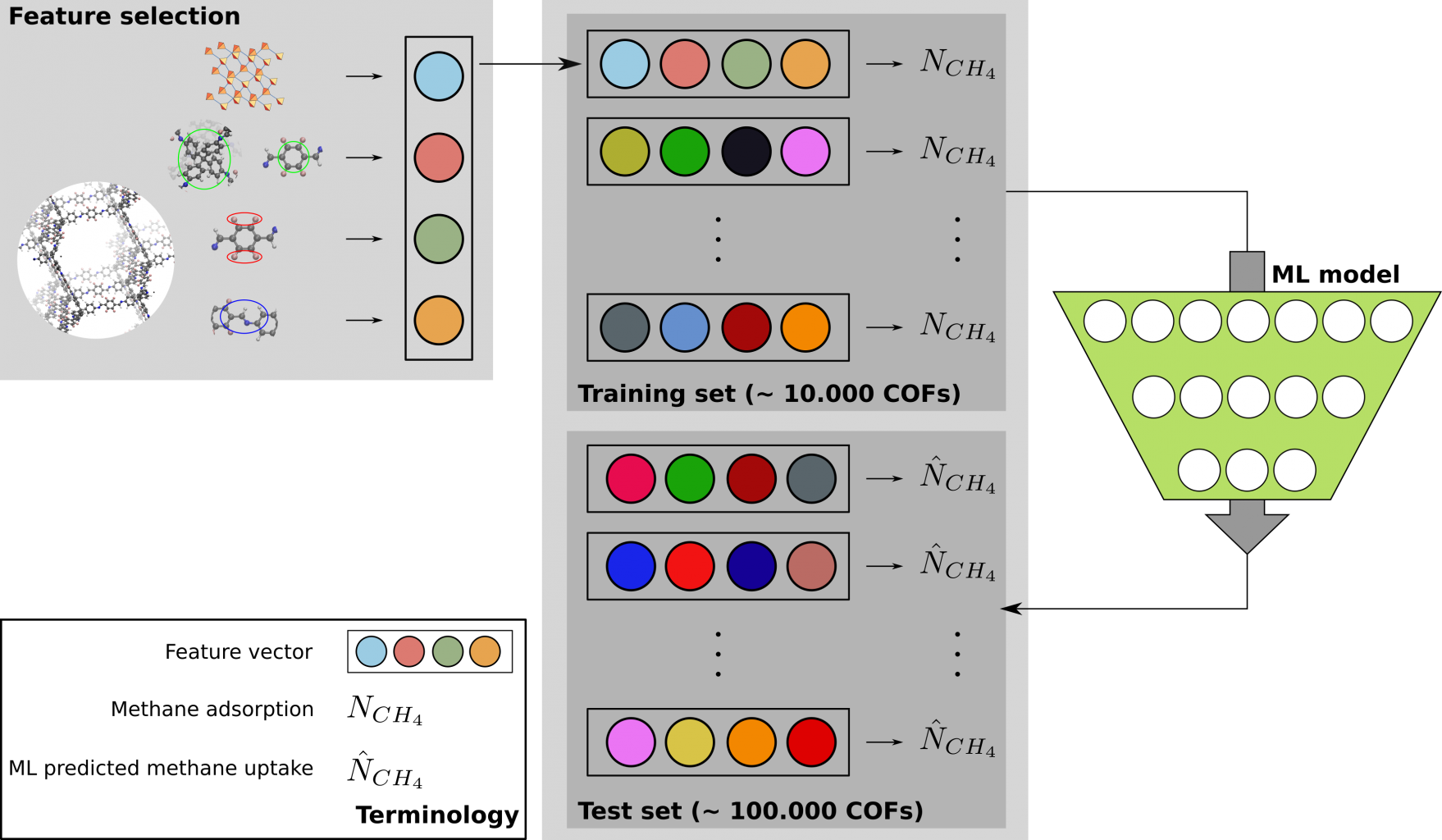
Goal
The final goal of this thesis is to identify those COFs within a diverse database that achieve the optimal gravimetric methane excess uptake. This will be realized in two steps. As an accurate description of the atomic interactions in each COF is needed, the first step will be to construct force field models for the periodic structures that are present in the database. These will be derived from the cluster models of the individual components from which the structure is generated, taking into account the natural decomposition of COFs in the three aforementioned domains: core, linkage and functional groups. For each of these elements, a domain-specific cluster force field will be assembled by carefully comparing the parameters obtained from quantum mechanical reference data when it is placed in different molecular environments [7]. The so-generated force field parameters for each component are finally combined to obtain an accurate system-specific description for each of the materials that can possibly be generated.
Secondly, a database will be proposed by selecting a list of possible topologies, cores, linkages, and functional groups and screened for the gravimetric excess methane uptake using Grand Canonical Monte Carlo (GCMC) simulations. Several machine learning algorithms, such as nonlinear support vector machines, decision trees, and neural networks, will be checked on their ability to predict the methane capacity using multiple feature classes. Features based solely on the individual components can vary from just indicating which domain element is chosen, to characterizing these elements thoroughly, for example using Coulomb matrices or autocorrelation descriptors. Finally, also structural properties such as pore diameter and void fraction will be included. Potentially, an additional layer can be included that predicts these structure properties from the individual components. The ability and relative importance of each feature to predict the COF performance will be carefully checked by making use of the SHAP method.
The student will be actively coached to make him/her acquainted with the advanced simulation techniques, early in the thesis year, and to transfer necessary programming skills needed to perform the research.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsNanoporous materials, Covalent organic frameworks, database screening, machine learning, methane storageReferences
[1] S.-Y. Ding and W. Wang, "Covalent organic frameworks (COFs): from design to applications", Chem. Soc. Rev., vol. 42, no. 2, pp. 548-568, 2013.
[2] O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, "Reticular synthesis and the design of new materials", Nature, vol. 423, pp. 705-714, 2003.
[3] Y. J. Colón and R. Q. Snurr, "High-throughput computational screening of metal-organic frameworks", Chem. Soc. Rev., vol. 43, no. 16, pp. 5735-5749, 2014.
[4] R. Mercado, R.-S. Fu, A. V. Yakutovich, L. Talirz, M. Haranczyk and B. Smit, "In silico design of 2D and 3D covalent organic frameworks for methane storage", Chem. Mater., vol. 30, no. 15, pp. 5069-5086, 2018.
[5] S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, "Understanding the diversity of the metal-organic framework ecosystem", Nat. Commun., vol. 11, 2020.
[6] M. Fernandez and T. K. Woo, "Large-scale quantitative structure-property relationship (QSPR) analysis of methane storage in metal-organic frameworks", J. Phys. Chem. C, vol. 117, no. 15, pp. 7681-7689, 2013.
[7] L. Vanduyfhuys, S. Vandenbrande, J. Wieme, M. Waroquier, T. Verstraelen, V. Van Speybroeck, “Extension of the QuickFF force field protocol for an improved accuracy of structural, vibrational, mechanical and thermal properties of metal-organic frameworks”, J. Comput. Chem., vol. 39, no. 16, pp. 999-1011, 2018.
Contact
Veronique Van Speybroeck