Training a reactive (ReaxFF) force field for the simulation of zeolite nucleation in a hydrated silicate ionic liquid
Training a reactive (ReaxFF) force field for the simulation of zeolite nucleation in a hydrated silicate ionic liquid
Promotor(en): T. Verstraelen, J. Vekeman /25451 / Model and software developmentBackground and problem
Molecular dynamics (MD) simulations of complex chemical systems rely on the computation of Newton’s equations of motion to propagate the system through time. Afterwards, relevant properties of the system can be extracted through postprocessing the obtained data using statistical mechanics. To drive MD simulations, forces between atoms need to be computed which is often done through a set of parameters (the so-called force field) describing the interactions within the system of interest. One such forcefield is ReaxFF that relies on hundreds of parameters derived empirically by fitting to accurate reference data. [1] Because of the size of the parameter space, the fitting of this force field is non-trivial and at the Center for Molecular Modeling a number of tools are being developed to speed up this process, such as very efficient optimizers as well as methods to extract more training data from electronic structure calculations.
An area where reactive molecular dynamics could offer important insights, is the field of zeolite formation. Indeed, although zeolites are very important for many industries, their formation is still not fully understood due to the difficult experimental conditions in which formation occurs. On the other hand, theoretical models have been hampered by the troublesome modeling of hydrogen bonds present in the crucial water solvent. Recently, a new synthesis route was proposed whereby the main problems for direct observation and characterization – pressure build-up and gel-formation – are evaded by using hydrated silicate ionic liquids (containing silicic acid, water, Al(OH)3 and a cation such as sodium). [2] Aside from experimental investigations, the modeling of the ionic liquid and first nucleation steps of the zeolite is crucial as it is the only way to offer an accurate view on the process at a molecular level. Fortunately, the low amount of water, the presence of only small molecular (alumino)silica species – such as monomers, dimers, 3- and 4-rings – and the fact that formation only starts upon introduction of low amounts of aluminum, allow construction of relevant molecular models.
Goal
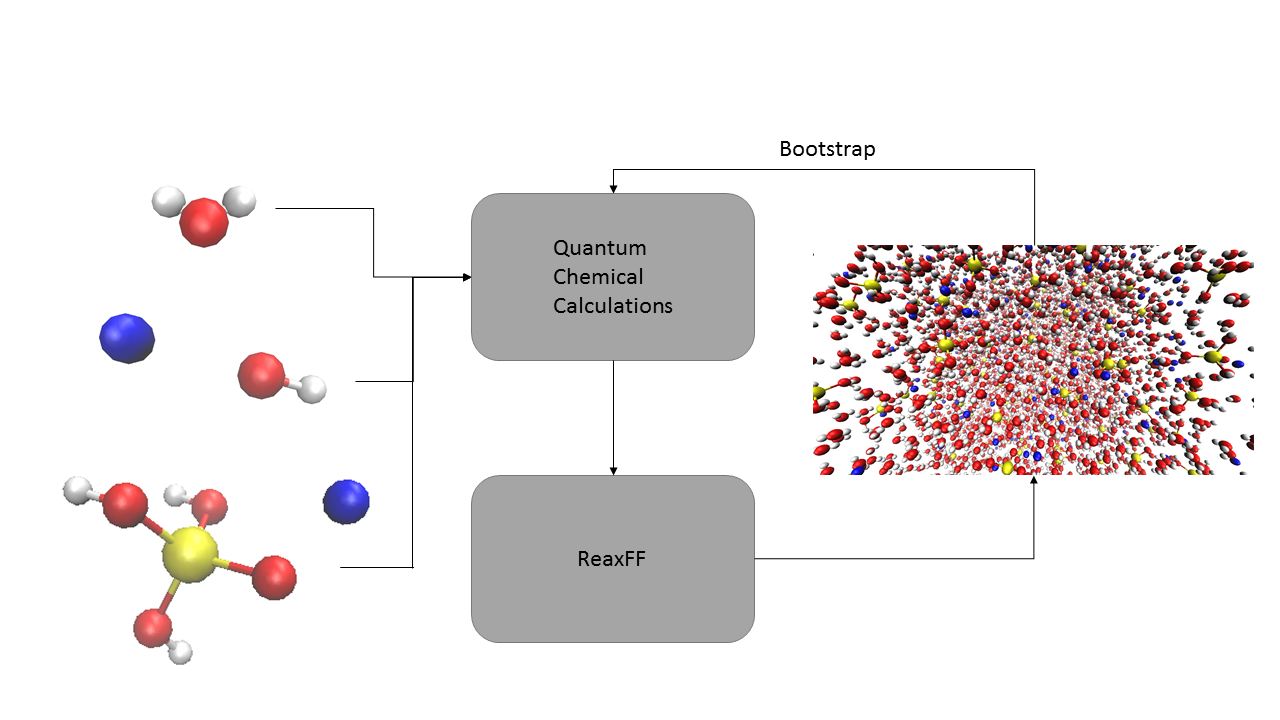
The goal of this thesis is then firstly to generate the necessary data for an appropriate training of the ReaxFF force field specifically for the hydrated silicate ionic liquid system of interest. More specifically, the data will consist of quantum chemical calculations, including equations of state, energy barriers, interaction energies and dissociation energies. When generating data, the chemical reaction that is considered most important for the process will be given priority, namely the deprotonation of silicic acid and the resulting proton transfer to/in water, as it is needed to describe the dynamics of the ionic liquid properly. In a second stage, the available data will be used to efficiently train the ReaxFF force field using the available tools at the Center for Molecular Modeling. In order to speed up this process, the training data will be enriched through bootstrapping. More specifically, equally-spaced snapshots will be taken from the simulation trajectories and single-point DFT calculations will be performed to obtain the forces which will be reintroduced in the fitting algorithm. Thirdly, trial molecular dynamics simulations of the HSIL system will be performed to validate the used training set by benchmarking macromolecular observables against experimental data from collaborators at KU Leuven.
List of figures:
Figure 1: A schematic overview of the project: quantum chemical calculations of simple systems are used to fit the ReaxFF force field. Then, molecular dynamics simulations are run and bootstrapped to further optimize the parameters.
- Study programmeMaster of Science in Civil Engineering [EMCIVI], Master of Science in Physics and Astronomy [CMFYST]KeywordsReaxFF, Reactive Force Field, Training, Molecular dynamicsReferences
[1] T. P. Senftle, S. Hong, Md Mahbubul Islamn, S. B. Kylasa, Y. Zheng, K. Kyung Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. Metin Aktulga, T. Verstraelen, A. Grama and A. C. T. van Duin, npj Computational Materials, 2016, 2, 15011
[2] van Duin, A. C. T.; Dasgupta, S.; Lorant, F.; Goddard, W. A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396−9409
Contact
Toon Verstraelen