Towards accurate estimates for transition rates of physical transformations in nanoporous crystals
Towards accurate estimates for transition rates of physical transformations in nanoporous crystals
Promotor(en): V. Van Speybroeck, L. Vanduyfhuys /20NANO06 / Nanoporous materialsProbleemstelling:
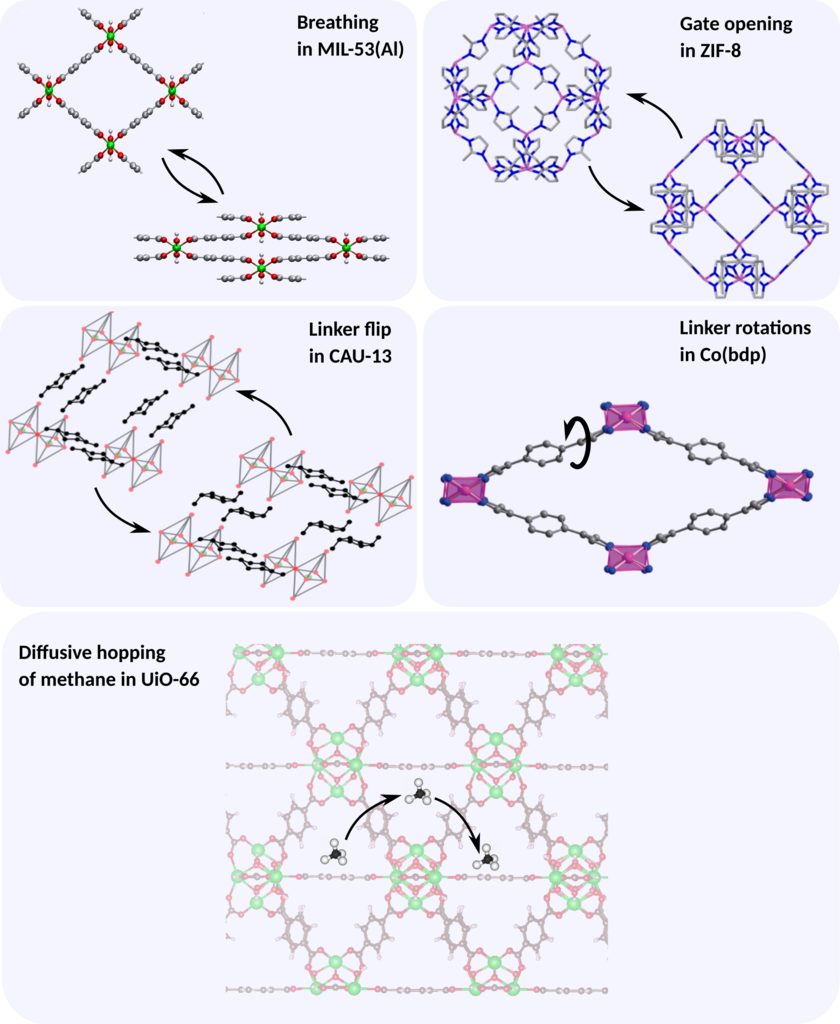
Metal-organic frameworks (MOFs) are a class of porous crystalline materials that consist of inorganic metal nodes connected with organic linkers. These hybrid structures are interesting materials from an application point-of-view due to their versatility, making them ideal candidates for usage in gas separation, adsorption or storage, drug delivery, sensing applications… Moreover, the connection between organic and inorganic parts may offer intrinsic flexibility, which gives rise to exceptional phenomena such as gate-opening effects and framework breathing. With an eye toward industrial applications, it is crucial to know how fast these physical processes progress. Recent experimental advances have enabled to monitor several physical transformations either through time resolved XRD measurements or via NMR spectroscopy. From these measurements, it becomes possible to estimate the transformation rate within the system. Unfortunately, these experiments are both time-consuming and expensive. Therefore, theoretical input is required to predict transformation rates on-the-fly.
The theoretical estimation of such rate constants is the subject of transition state theory. Herein, one represents the process in terms of a transition state (TS), which represents an intermediate state in between the initial (I) and final (F) state. In terms of the Helmholtz free energy surface, the initial and final states are represented by local minima, while the transition state is given by a saddle point, which is a maximum in the direction of the process progression and a minimum in the remaining perpendicular directions. This direction of the process progression plays a crucial role and is quantified by a so-called collective variable (CV), which is a mathematical function of the molecular degrees of freedom (i.e. the atomic positions). In order to predict transition rates, the free energy profile as a function of the CV needs to be determined via advanced molecular simulations. However, for many processes, the choice of the collective variable is not unique. For that purpose, a protocol was developed at the Center for Molecular Modeling that estimates the transformation rate independent of the choice of CV:
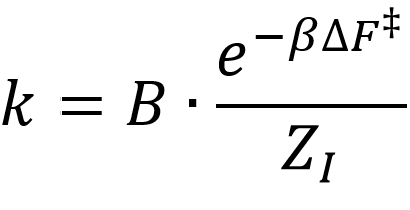
Herein, ΔF represents the difference in free energy between the transition state maximum of the FES and initial state minimum, ZI represents the partition function of the initial state and takes into account the entire valley in the free energy profile corresponding with the initial state, while B represents a more complex pre-exponential function, which takes the width of the transition state into account.
Doelstelling:
In this thesis, we will first apply the proposed methodology to predict phase transformation rates in breathing MOFs. Upon breathing, the unit cell volume changes significantly going from a closed pore phase to a large pore phase or vice versa (see figure). Consequently, a straightforward choice for the CV of this transformation is the unit cell volume. However, many processes cannot be described by means of a single CV, but require 2 CVs with a corresponding 2D free energy surface. Therefore, the second goal of this thesis will be to investigate how the expression for the transition rate can be extended towards 2D free energy surfaces. This part will involve more theoretical derivations within the field of statistical physics and molecular simulation. For that purpose, different physical processes will be considered, such as the linker flip and linker rotations represented in the figure.
To predict transformation rates within breathing MOFs, we will focus on DUT-8, consisting of 2D layers with metal paddle wheel units connected by naphthalenedicarboxylate linkers. These 2D layers are held together by DABCO linkers, which serve as pillars. The calculation of transition rates requires the construction of the FES using advanced molecular simulations. For that purpose, molecular dynamics (MD) simulations will be performed, in which Newton’s equations of motion are integrated over time, using a classical force field to describe the molecular interactions. By means of thermodynamic integration of the mechanical pressure, the free energy can then be derived as a function of the unit cell volume, i.e. the collective variable in this case, which eventually leads to the transition rate.
Besides a direct comparison of the theoretical phase transformation rate with experiment, additional spectroscopic characterization can be performed to assess the validity of the model system. XRD patterns and NMR parameters can be obtained from snapshots of the MD simulations. Calculation of the latter has become possible since the recent development of the GIPAW approach, which requires computationally demanding ab initio simulations. The theoretical XRD and NMR results will be compared with the available time-resolved experimental data.
As the building block concept in MOFs allows to easily replace metal and linker units, it is of interest how a substitution of the linker and/or metal atom changes the kinetics of the phase transformation. The relative trends in the theoretical phase transformation rate following metal substitution or linker functionalization will be predicted and, where possible, compared with literature. As a result, the final aim is to significantly increase the understanding of the dynamics of various transitions in MOFs, and to be able to apply the newly gained knowledge towards the functional design of new materials.
This thesis consists of two major components: a theoretical component and a computational component. The focus can be shifted toward one component or the other depending on the interest of the student.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsHelmholtz free energy, Transition rate, Transition state theory, Metal-organic frameworks, X-Ray Diffraction, nuclear magnetic resonance
Contact
Veronique Van Speybroeck