Thermodynamic modeling of reactive and associating molecular systems
Thermodynamic modeling of reactive and associating molecular systems
Promotor(en): T. Verstraelen, A. Ghysels /MM_14_MODEV_09 / Model and software developmentMolecular dynamics (MD) and Monte Carlo (MC) simulations are powerful techniques for the modeling the thermodynamics of molecular systems. With MD, given an energy function that describes the forces between all the atoms of the system, the positions of the atomic nuclei evolve in time by integrating the Newton’s equations of motion. The MC method, on the other hand, relies on equilibrium statistical mechanics. Here, random displacements of the atomic nuclei are tried and accepted according to the Metropolis acceptance criterion to obtain canonical distributions. Force-field based MD or MC simulations are a very attractive alternative to ab initio simulations because of the small computational cost associated with the evaluation of atomic forces, which allows the simulation of much larger systems and timescales. Classical force fields, however, are limited to weakly interacting systems, because atomic bonds cannot be formed or broken during the simulation. This limitation may be overcome with the reactive canonical Monte Carlo (RCMC) method [1], which makes it possible to simulate important chemical interactions between molecules such as association (hydrogen bonding, charge transfer) and proton transfer.
Goals In this thesis, the RCMC method will be implemented and tested in the in-house force field based molecular dynamics package of the CMM. The key challenge is here the integration with the conventional MD routines. The implemented RCMC code can be applied to a variety of interesting physico-chemical processes. As a first important application, the formation of protonated methanol clusters in acidic nanoporous zeolites will be studied, as illustrated in Figure 1. These clusters have been shown to play a key role in the industrial methanol-to-olefin process.
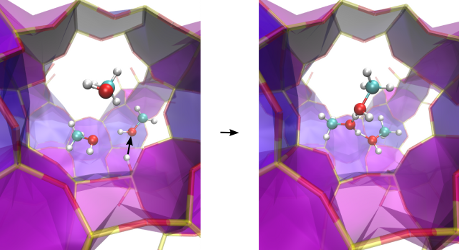
Figure 1. Illustration of proton transfer from the acid site of a zeolite to three methanol molecules, forming a protonated cluster.This master thesis is challenging and focuses on programming and testing of new algorithms. This topic is ideally suited for a student who has a strong interest in statistical physics.
[1] Johnson K. Reactive canonical Monte Carlo: A new simulation technique for reacting or associating fluids. Molecular Physics, 1994, 81, 3, 717-733.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsMolecular modeling, Monte Carlo, Molecular dynamics, Statistical physicsRecommended coursesSimulations and Modeling on the Nanoscale, Molecular Simulations of Biosystems
Contact
An Ghysels
Toon Verstraelen