A thermodynamic approach to efficient sampling of the unit cell shape for flexible crystals
A thermodynamic approach to efficient sampling of the unit cell shape for flexible crystals
Promotor(en): A. Ghysels, V. Van Speybroeck /15_MODEV13 / Model and software developmentThe discovery of metal-organic frameworks (MOFs) was a milestone in material science. These hybrid materials consist of metal-oxide clusters interconnected by means of organic linkers, resulting in a 3D periodic framework with nanosized pores. Due to the porosity, these nanoporous crystals show many promising applications such as carbon dioxide capture, hydrogen storage and the separation and detection of gasses. Moreover, some frameworks are inherently very flexible, enabling them to change the size and shape of their unit cell under influence of external stimuli, such as temperature, pressure, or adsorption of guest molecules. The MOF material MIL-53, for instance, can shrink its volume by about 40% while retaining its crystallinity. This special type of flexible behavior is known in literature as framework breathing. This property opens a new range of applications such as nano dampeners and nano springs. Due to the large variety in metal oxides and organic linkers, a large number of MOF materials can be synthesized. Computational research may assist in selecting those MOFs that are the best candidate materials for applications.
Recently, a thermodynamic model has been proposed at the Center for Molecular Modeling to describe the breathing behavior of metal-organic frameworks under influence of adsorption of guest molecules. This model is able to predict the framework transformations (breathing) stimulated by external mechanical pressure or by adsorption of guest molecules. A crucial part of the model is the free energy profile of the empty MOF framework, which can be obtained from advanced molecular simulations. At present, this free energy is constructed as a one-dimensional profile, i.e. as a function of the volume only. The most stable structure is then found at the minimum of this free energy curve.
However, in reality the crystal structure is defined by its unit cell shape as well, which yields a six-dimensional free energy surface. Figure 1 illustrates the six dimensions in the unit cell (length of cell vectors and their angles). The current limitation of the six-dimensional surface to a one-dimensional profile forms a severe restriction to the present thermodynamic model. The thermodynamic model needs to be extended to cover all six dimensions, and an advanced molecular simulation algorithm needs to be designed to sample all dimensions. The knowledge of the free energy profile as a function of all shape parameters would allow for the exact prediction of the most stable crystal structure at a given temperature, and could thus help understand the breathing behavior. Figure 2 gives an example for a two-dimensional surface of the MIL-53 material.
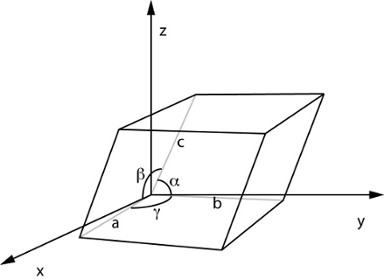
Figure 1: The unit cell has six dimensions: one for the unit cell volume, five for the unit cell shape.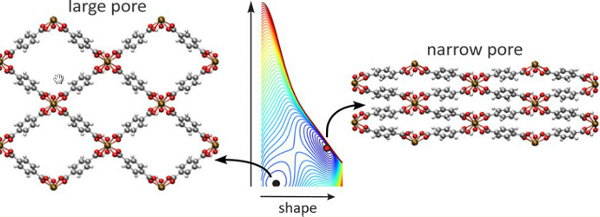
Figure 2. Two-dimensional free energy landscape of MIL-53 material: the most stable structure is here the narrow pore shape, not the large pore shape.Goal
Predicting equilibrium crystal structures at a given temperature and pressure is an import problem in different fields of science. We are particularly interested to construct multi-dimensional free energy profiles for nanoporous materials which exhibit framework flexibility. Recently, several advanced simulation techniques were implemented at the CMM to obtain one-dimensional free energy profiles. The goal of this thesis is twofold. First, the student will further extend the advanced simulation techniques in order to obtain high-dimensional free energy profiles. For this goal, the student will need to get familiar with standard molecular dynamics (MD) simulation techniques, where Newton's equations of motion are integrated. Additionally, the student will explore how steered dynamics can improve statistics, where the simulation is biased towards sampling less populated regions of the free energy profile, followed by corrections to undo the bias. Sampling of the unit cell shape is here a completely new aspect in the field. Second, the student will extend the thermodynamic model and apply the advanced simulation techniques to MOFs to predict their equilibrium crystal structure from simulations. The work performed by the student will contribute to the thermodynamic model developed and used by the researchers at the CMM and other research groups.This topic is on the cross road between statistical physics, material engineering and molecular simulation. On the one hand you will need to understand the advanced simulation techniques in order to extend them. Understanding this type of simulations requires good understanding of statistical physics. On the other hand, communicating your results will make you comfortable with material science. The topic will deepen your knowledge about simulation techniques and computational physics. If needed, the required programming and simulation skills will be transfered during the thesis.
Physics aspect: statistical physics are used to make the connection between sampling at microscale and the observable macroscale behavior, concepts of ergodicity, degrees of freedom.
Engineering aspect: rendering computational sampling methods more efficient in order to ensure complete sampling, application of theory to real materials.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) nano, modelingKeywordsMolecular simulation, Free energy calculations, Crystals, Nanoporous materials
Contact
An Ghysels
Veronique Van Speybroeck