Study of the dissociation of diatomic molecules with advanced electronic structure methods
Study of the dissociation of diatomic molecules with advanced electronic structure methods
Promotor(en): D. Van Neck, T. Verstraelen /MM_13_FUND06 / Model and software development, Many-particle physicsProblem A matrix product state (MPS) is a relatively recent wavefunction ansatz that allows a systematic convergence of the solution of the electronic Hamiltonian towards the full configuration interaction (FCI) limit at a reduced computational cost. It was shown to be very effective for the prediction of the energy and the reduced two-particle density matrix (2DM). A new implementation of the MPS method is developed at the Center of Molecular Modeling that enables very accurate computations on small systems (up to 40 electrons in 40 orbitals).
One of the useful applications of this new MPS program is to study the dissociation of diatomic molecules. A high quality reference data set with properties of the diatomic molecules along the dissociation path (total energy, charge transfer energy, linear response, bond order, ...) is currently not available, while such data are very useful for the development of reactive and polarizable forcefield models. Single reference methods, like Hartree-Fock, can only describe the stable diatomic and the fully dissociated state but not the transition between the two.
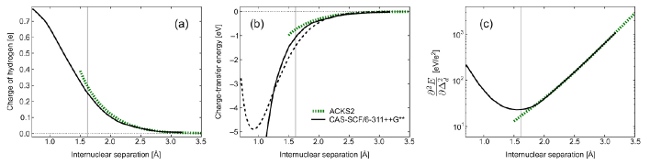
Figure: dissociation of the HF molecule at the CASSCF/ 6311++ G** level compared with the empirical ACKS2 model. (a) charge transfer from F to H. (b) charge transfer energy. dashed black = total CASSCF. full black = CASSCF. green dotted = ACKS2. (c) bond softness. Goal In this thesis, the MPS method will be used as FCI solver in a complete active space self consistent field (CASSCF) loop, where the correlation is fully resolved for a number of orbitals (at MPS (FCI) level), in an approximate environment kept at Hartree-Fock level. The SCF part ensures that the Hessian is positive definite for all possible orbital rotations. From the CASSCF solution, a 2DM can be extracted, which allows for an easy evaluation of expectation values. A database of dissociation properties of homo and heteronuclear diatomics with elements up to krypton will be studied with the MPS program. When possible, the results are compared with CCSD(T). The application of constraints on the wavefunction enables the study of the charge transfer contribution to the binding energy and the chemical softness of the bond. Also the partial charges and the chemical bond order can be derived at each internuclear distance. The correlation between these parameters will be studied and it will be tested to what extent these results are compatible with the ACKS2 model.
More info:
MPS: http://arxiv.org/abs/1008.3477
Questions are always welcome: sebastian.wouters@ugent.be

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsMatrix product states, Diatomic molecules, Chemical bonding, Electronic linear responseRecommended coursesManybody physics (C001759); Computational physics (C001827)
Contact
Dimitri Van Neck
Toon Verstraelen