Structural characterization of solvents confined in zeolites
Structural characterization of solvents confined in zeolites
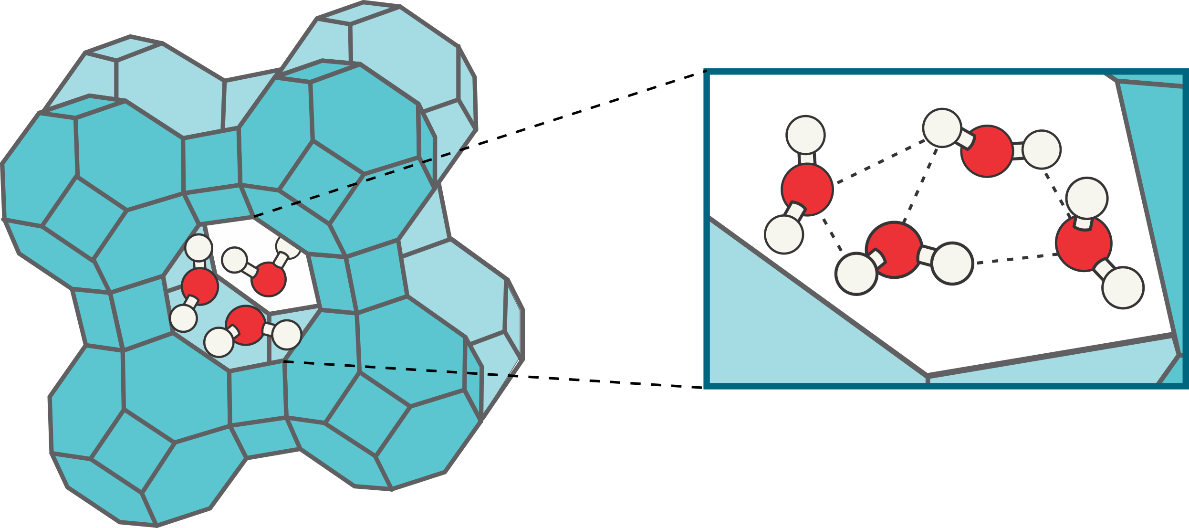
Promotor(en): V. Van Speybroeck /18MODEV11 / Model and software development, Nanoporous materialsWithin the past few decades, the exponential growth of powerful computing resources has attributed to the increasing importance of computational physics, allowing to simulate larger systems at an increasingly higher accuracy. Within the field of molecular modeling, the class of nanoporous materials [1], including zeolites [2] and metal-organic frameworks, has received a great deal of attention and has most certainly benefited from these recent advances in the field of computational physics. Zeolites make up a very interesting class of nanoporous materials, which can be found in nature, but can also be synthetically designed for specific applications, such as gas purification, catalysis, and removal of radioisotopes from radioactive waste streams. Structurally, zeolites can be described as crystalline aluminosilicate frameworks composed of AlO4/SiO4 tetrahedra that are connected to one another via O atoms, with extraframework cations to compensate for the negative charge of the aluminum complexes. In the pores of the zeolitic framework, a wide variety of physical and chemical processes can take place, in which the presence of a solvent often plays an important role. Typical solvents such as water and methanol are in itself already difficult to model, due to the decisive influence of hydrogen bond formation upon the liquid’s structure, such that modeling their presence within nanoporous materials poses an even bigger challenge. Despite several experimental and computational indications that the confinement of solvent molecules within nanoporous materials yields significant modifications in the structural arrangement of the solvent molecules, a thorough computational study of the phenomenon has not been performed to date.
Goal
In this thesis, a complete structural characterization of different solvent molecules confined within zeolitic frameworks is aimed for. Starting from bulk solvent properties as benchmarking results, the effect of confinement will be investigated for different solvent loadings of the zeolite using ab initio molecular dynamics (AIMD). Within the technique of AIMD, the electronic Schrödinger equation is solved numerically for every configuration of the structure’s nuclei, which are evolved throughout time by means of Hamilton’s equations of motion. However, as a numerical integration of Hamilton’s equations of motion results in a sampling of the microcanonical (NVE) ensemble, extensions of the Hamiltonian description are necessary to be able to also sample the canonical (NVT) and isothermal-isobaric (NPT) ensembles, corresponding with actual, physical operating conditions.
For bulk solvents, a clear distinction can be made between the liquid and gas phases, whereas for confined solvents the identification of the state of matter becomes more difficult due to the limited amount of absorbed molecules, which complicates a classification in terms of the macroscopic ‘liquid’ and ‘gas’ notions. Therefore, the structural properties of different solvents, including water, methanol, and ethanol, will be investigated using as many structural identifiers as possible, to elucidate the structural changes and similarities between the confined and bulk solvent states [3], at relevant operating conditions. Possible structural identifiers include radial distribution functions (RDFs), X-ray diffraction (XRD) patterns, and hydrogen bond statistics, which might need to be complemented by other structural identifiers in order to obtain a fully structurally discriminative description of the confined and bulk solvent states. Complementary information with respect to the accuracy of modeling can furthermore be provided by comparison with experimental data, supplied by the COK, with whom the CMM has established a close scientific collaboration during the past few years. Finally, a preliminary screening of the influence of nuclear quantum effects (NQEs) can also be pursued by making use of the so-called generalized Langevin equation (GLE) thermostat, which modifies the MD sampling in order to account for the quantum effects inherent to the atomic nuclei, an effect which can be of major importance in accurately describing the hydrogen bond network.
Engineering and physics aspects
Engineering aspect: advanced molecular modeling of solvents in nanoporous materials
Physics aspect: elucidating molecular confinement effects using structural identifiers

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELINGKeywordsNanoporous materials, molecular simulations, confined solvents, hydrogen bonding, Nuclear quantum effectsReferences
[1] A. G. Slater and A. I. Cooper, Science, vol. 348, no. 6238, 2015
[2] V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell, and C. R. A. Catlow, Chem. Soc. Rev., vol. 44, no. 20, p. 7044-7111, 2015
[3] C.-H. Wang, P. Bai, J. I. Siepmann, and A. E. Clark, J. Phys. Chem. C, vol. 118, no. 34, p. 19723-19732, 2014
Contact
Veronique Van Speybroeck