Simulatie van het osmotisch ensemble voor zeolieten
Simulatie van het osmotisch ensemble voor zeolieten
Promotor(en): A. Ghysels, V. Van Speybroeck /MM_13_MODEV09 / Model and software development, Nanoporous materialsNanoporous zeolite crystals are widely used for the adsorption and separation of small molecules, such as CO2 and volatile organic compounds. In recent years, many structural and chemical variants have been proposed to widen the adsorption range and selectivity of these zeotype structures. Zeolites are also known to possess a certain degree of flexibility, depending on the framework type. The effects of framework flexibility on the adsorption properties are still not well understood.
An important question is which zeolite material is optimal in terms of adsorption and selectivity. The optimal zeolite candidate should be selected from the extensive library of (hypothetical) structures. However, the synthesis of all possible variants is very time consuming if at all possible. Molecular modeling of these materials could provide an estimation of the adsorption capacities and selectivities of nanoporous materials before they are made.
At the Center for Molecular Modeling, we are actively developing molecular mechanics force fields and molecular simulation algorithms to efficiently yet accurately describe the structure and dynamics of zeolites and other porous materials such as metal organic frameworks. Simulation of the adsorption process remains an issue, because the appropriate ensemble is not at hand.
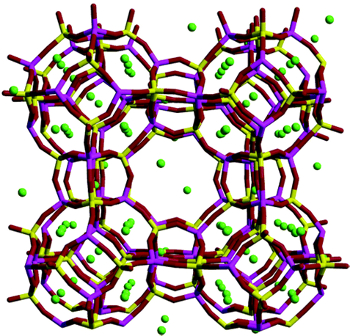
Zeolite structure with adsorbed guest molecules (green). In molecular dynamics, the behavior of a molecular system is followed over time by integrating the Newton equations of motion. In an adsorption experiment, however, the number of guest molecules may fluctuate when molecules enter and leave the pores. This is clearly not possible in a standard constant NVT ensemble, where the number of atoms in the system is kept fixed. The preferred ensemble for studying adsorption is the osmotic ensemble, where the number of small molecules inside the nanopores and the volume of the porous host are allowed to fluctuate, while the pressure and temperature are kept fixed.
In this thesis, sampling of the osmotic ensemble will be implemented and tested in our in-house force field based molecular dynamics package. The key challenge is here that insertion of deletion of a guest molecule disturbs the set of Newton equations. Once this algorithm is implemented, it will be used to predict the adsorption isotherms of small molecules in zeolites. In addition, the influence of adsorbed molecules on the pore volume will be studied. A possible extension is the constant pH ensemble, relevant to study reactions in zeolites with catalytic Brønsted acid sites.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsAdsorption, Framework flexibility, Statistical physicsRecommended coursesComputational physics (C001827); Modeling and simulation at the nanoscale (E023370)
Contact
An Ghysels
Veronique Van Speybroeck