Screening potentially interesting covalent organic frameworks by accurate force field simulations
Screening potentially interesting covalent organic frameworks by accurate force field simulations
Promotor(en): V. Van Speybroeck /16MODEV10 / Model and software developmentNanoporous materials are inherently present in several daily applications such as energy storage, gas separations, CO2 capture, … . They can be categorized either into microporous (pore diameter less than 2 nm) or mesoporous materials (pore diameter from 2 to 50 nm), and are tractable due their high surface area, porosity and chemical versatility. Experimentally, new materials are being synthesized at a considerable pace, using the most advanced methods to tailor materials at the nanometer scale level. Model-guided design is an essential part in this overall process to guide the experimentalists, to discover new applications and to optimize current processes to their full potential.
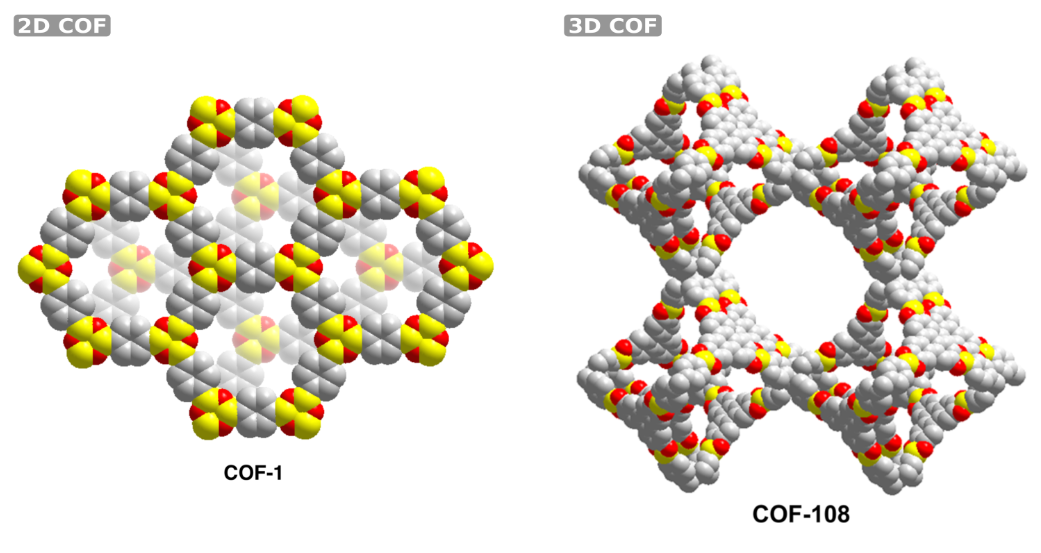
Covalent organic frameworks (COFs) form a recently proposed class of crystalline materials, consisting solely of organic moieties connected by strong covalent bonds [1]. To describe these COFs, they are often divided into secondary building units. In Figure 1, it is shown how the stacking of these building units may lead to either 2D or 3D COFs. 2D COFs are composed of stacked layers, resulting in dense structures and one-dimensional channels, whereas 3D COFs are expected to be highly porous, with low densities and high surface areas. The absence of metals in COFs makes them much lighter than their sister materials, MOFs and zeolites, while still retaining a high surface area and pore volume. These properties make them tractable towards applications such as adsorption, catalysis, light harvesting and photocatalytic activity, where the material should be as light as possible.
Thanks to the large amount of potential building blocks, the variety of possible topologies to build the scaffold, and the ability to be post-functionalized with complexes introducing extra active functionalities to the material, a large versatility of COFs arises. Ideally, the various degrees of freedom in constructing these materials may be controlled and fine-tuned at the molecular level, yielding materials with the proper structural, electronic and functional characteristics for a given application. The exploration of COFs, both experimentally and computationally, is now fully in expansion.
Goal
The Center for Molecular Modeling has ample experience in modeling nanoporous materials such as zeolites and metal-organic frameworks, and we decided to extend our modeling efforts towards COFs. As the fields of COFs is largely unexplored today, it is the intention of this topic to perform a screening study on potentially interesting COFs starting from compelling building blocks, in close synergy with several experimental partners. The aim of this topic is to select those COFs with desired structural features (i.e. pore size distribution, window size…), mechanical properties (bulk modulus, degree of sliding in 2D COFs), and thermal properties (thermal expansion, temperature dependence of the bulk modulus). In particular, we are interested in COFs that are stable and rigid enough to avoid collapse of the framework under given temperature and pressure conditions. In other COFs, we want to anchor metal complexes which might be interesting for catalysis. It is, however, essential to control the pore size for these applications. Moreover, for 2D COFs, the layers should not be able to easily slide one over the other. Such sliding would decrease the effective pore size and block the access to the metal complexes for the reaction species.
The screening study will start from certain building blocks which may then be assembled towards various COFs with different topologies. At first instance, the most prevalent topologies exhibiting the highest symmetry will be selected, but an extension towards other topologies for designed pore sizes might be necessary. We will make use of the Zeo++ software designed for the high-throughput analysis of porous materials [2]. After having obtained viable initial structures, a first screening based on textural properties will be performed, which will yield information on structural features such as the pore diameter, accessible volume area, void fraction, pore size distribution. At second instance, the thermal and mechanical properties of interest will be determined using a range of simulation approaches such as advanced molecular dynamics techniques, Monte Carlo simulations, normal mode analysis and geometry optimizations, which were all previously tested at the CMM. However, due to the broad set of target materials, it is impossible to use full quantum mechanical calculations. Therefore, we will rely on dedicated force fields, since similar approaches for metal-organic frameworks at the CMM yielded satisfactory results.
A force field is a mathematical expression of the potential energy of the molecular system, without explicitly considering the electronic structure of the material. Force fields may give reliable structural data provided they are constructed in a robust way using accurate benchmark data. The CMM has developed a force field protocol, QuickFF, which is designed for a quick and easy derivation of force fields for metal-organic frameworks based on quantum mechanically derived benchmark data [3]. Within the framework of this thesis subject, we will extend the QuickFF protocol towards COFs. For COFs, only a few force fields are available, moreover they are limited to boron-based COFs. In general, the force field consists of two contributions: one describing the covalent interaction between bonded atoms, and one describing the non-covalent interaction between non-bonded atoms. The force fields are derived to reproduce reference quantum mechanical based reference data.
In this thesis, the student will perform quantum mechanical calculations to generate reference data required for the force field derivations. Next, QuickFF will be applied to derive the force field. Once these force fields are available, several molecular simulations will be performed to compute all the required properties, which can be validated based on periodic, quantum mechanical VASP calculations currently carried out at the CMM. Finally, the student will use all results to present a complete characterization of the materials and propose which ones are promising for the application at hand. The selected materials will be discussed with our experimental partners who may experimentally test the modeled properties. The work performed in this thesis bridges the gap between quantum mechanics and thermodynamics via the use statistical physics and computational physics. The programming skills will be transferred during the course of the year when required.
Aspects
Physics aspect: Application of advanced theories from physics such as quantum mechanics, thermodynamics and statistical physics.
Engineering aspect: Complete mechanical and structural characterization of COFs to identify promising candidates for a given application.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELINGKeywordsCovalent organic framework, mechanical stability, Flexibility, layer stacking, molecular simulationsReferences
[1] P. J. Waller, F. Gándara and O. M. Yaghi, "Chemistry of Covalent Organic Frameworks," Acc. Chem. Res., vol. 48, no. 12, pp. 3053-3063, 2015.
[2] T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Mez and M. Haranczyk, "Algorithms and Tools for High-throughput Geometry-based Analysis of Crystalline Porous Materials," Microporous Mesoporous Mater., vol. 149, no. 1, pp. 134-141, 2012.
[3] L. Vanduyfhuys, S. Vandenbrande, T. Verstraelen, R. Schmid, M. Waroquier and V. Van Speybroeck, "QuickFF: A Program for a Quick and Easy Derivation of Force Fields for Metal-Organic Frameworks from ab initio Input," J. Comput. Chem., vol. 36, no. 13, pp. 1015-1027, 2015.
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck