Rapid estimation of the pKa with atoms-in-molecules
Rapid estimation of the pKa with atoms-in-molecules
Promotor(en): T. Verstraelen, P. Bultinck /16MODEV08 / Model and software developmentThe experimental determination of the pKa of acid groups in supramolecular systems is very challenging when many different acid sites are present, all with slightly different pKa values. This is typically the case in proteins but also in porous materials, e.g. zeolites, where a (random) distribution of different acid groups coexists. Different factors play a role, which can be divided into local effects, e.g. the electrostatic potential at the acid side, and non-local effects, such as the accessibility of the acid group.
Ab initio molecular dynamics simulations, in which the thermodynamic cycle of the deprotonation reaction is explicitly constructed, can provide reasonable estimates of a pKa, within one unit. However, such simulations are already computationally very demanding when just one pKa needs to be computed. An alternative computational approach is to establish quantitative structure-activity relationships (QSAR) between the pKa and quantum-chemical properties of the molecule that are relatively easy to compute. Such a QSAR approach is also very attractive when trying to design a molecule to have a specific pKa, e.g. by altering organic moieties close to the acid group.
Recently, several new variants of the Hirshfeld atoms-in-molecules method were proposed, which partition (computed) molecular electron densities into atomic contributions. These methods allow one the easily characterize the charge distribution with just a few numbers (atomic charges, multipoles, ...). The results of an atoms-in-molecules analysis is ideally suited as input for QSAR models.
Goal
The goal of this thesis is to test the effectiveness of newly developed atoms-in-molecules methods at the Center for Molecular Modeling and the Ghent Quantum Chemistry Group for the rapid prediction of pKa's for a broad range of organic compounds. The development of such a QSAR model will be carried out with databases of various organic molecules for which the pKa is known from experiment. This model will be validated with recently computed and measured pKa's of pH-sensitive dyes. An example is shown in Fig. 1: when the pH changes from 5 to 2, the azo bond in ethyl orange gets protonated and the dye color changes from orange (473 nm) to red (508 nm).
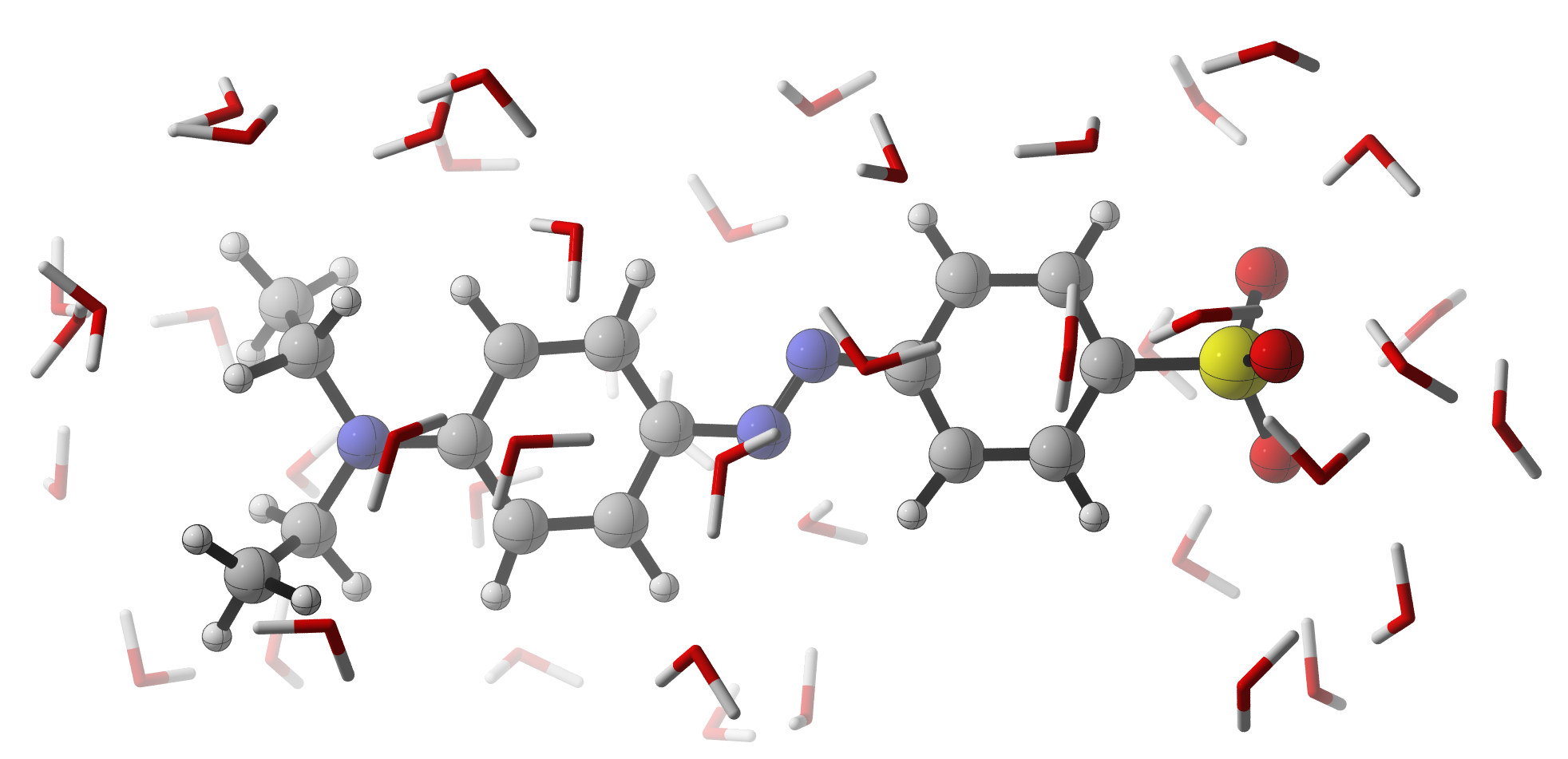
Fig. 1: Atomistic model of ethyl orange solvated in water.Once the QSAR model is validated, it will be used to design the pKa of pH-sensitive dyes by altering relevant functional groups. To obtain a useful color of the dye, i.e. in the visible part of the electromagnetic spectrum, the delocalized π-bond should remain extended over a large part of the molecule. Substituents on the aromatic system can be used to modulate the pKa. However, in order to embed the dyes into a polymer, additional substitutions are needed, which also have their impact on the pKa. All these effects have to be accounted for in the design of the dye.
Experimental and computational research on pH-sensitive dyes has been carried out at UGent in the groups of prof. Karen De Clerck and prof. Veronique Van Speybroeck, respectively. The application of the QSAR model in this thesis will be carried out in collaboration with these groups.

- Study programmeMaster of Science in Chemistry [CMCHEM]Keywordsquantitative structure-activity relationships, computational chemistry, pH sensitive deys, computational screening
Contact
Toon Verstraelen