Ranking molecular crystals with a many-body expansion
Ranking molecular crystals with a many-body expansion
Promotor(en): T. Verstraelen /25300 / Model and software developmentBackground and problem
Molecular crystals consist of a periodic stacking of molecules, for which the intermolecular interactions are relatively weak compared to the intramolecular ones. Still, the weak forces are strong enough, such that these crystals easily remain solid at room temperature. Molecular crystals are used in many industries, mostly in pharmacy, and also have promising applications in emerging technologies (e.g. organic semiconductors). For a single molecule, different metastable crystal phases exist, which are called polymorphs. See for example Fig. 1.
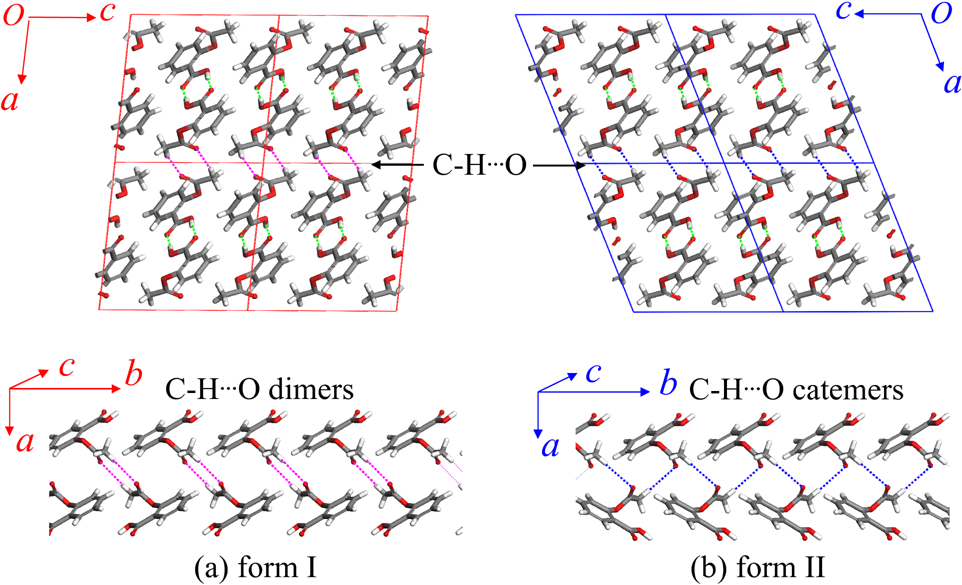
Figure 1. Polymorphs I and II of aspirin.
A fundamental challenge is to predict which metastable polymorphs could exist for a given molecule and this knowledge can make or break the development of a new drug or a new material. [1] Over the past decade, computational crystal structure prediction (CSP) has become a growing research discipline, in which a variety of molecular simulation techniques is used to find as many as possible polymorphs of a given molecular crystal. [2] An essential ingredient in this type of simulations is the computation of the stability of a hypothetical polymorph, which is governed by two main contributions: the weak interactions between the molecules and the strain of each molecule due to internal deformations.
Goal
Open source software packages will be used to compute post-Hartree-Fock approximations of the electronic energy, for configurations involving only a few molecules of the infinite crystal. Such calculations result in one-, two-, three-, ... body contributions to the total crystal polymorph energy in a many-body expansion:
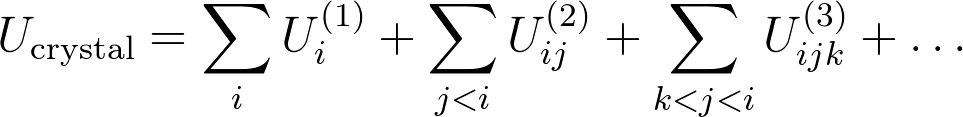
For molecular crystals, a truncation of this expansion after the third term should be reliable, but this hypothesis needs to be tested. Furthermore, from the second term, infinite lattice sums appear, which can only be computed up to a given cutoff distance. For the long-range interactions, an electrostatic model should be developed, which can be computed with Ewald sums. The development of a physically sensible model to compute this many-body expansion is the central challenge in this thesis.
In a CSP blind test, a set of drug-like molecules are published, whose crystal structures are known but kept secret. Research groups around the world are then invited to make predictions of their crystal structures, with typically about 20 groups participating. After the deadline of the blind test, all submissions are compared and an assessment is made of all methodologies used by the participants. All structures submitted in previous tests are publicly available, together with the most stable forms found in experiment. [2] These data will be used in this thesis to assess the accuracy of the model for the crystal polymorph energy.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]Keywordsintermolecular interactions, molecular crystals, many-body expansionReferences
[1] Beran, G. J. O. Modeling Polymorphic Molecular Crystals with Electronic Structure Theory. Chem. Rev. 116, 5567–5613 (2016). http://dx.doi.org/10.1021/acs.chemrev.5b00648
[2] Reilly, A. M. CSP Blind Tests. (2016). https://www.ccdc.cam.ac.uk/Community/initiatives/cspblindtests
Contact
Toon Verstraelen