Predicting the isotherms of adsorption through a combination of molecular simulations and thermodynamic models
Predicting the isotherms of adsorption through a combination of molecular simulations and thermodynamic models
Promotor(en): V. Van Speybroeck /17NANO12 / Nanoporous materialsNanoporous materials are structurally ordered materials that possess channels and pores of molecular dimensions, which makes them very tractable for various applications such as separation processes, gas storage, ... Popular examples of such materials include crystalline materials such as zeolites (inorganic materials consisting of Si, Al and O) and metal-organic frameworks (hybrid materials consisting of inorganic metal-oxides interconnected by means of organic linkers). Due to their nanometer-sized pores, they are able to adsorb large amounts of various guest molecules such as water, methane, carbon dioxide and others. As a result they can be used for gas storage, but also for gas separation, gas detection and catalysis of chemical reactions. In all of these applications it is essential to have knowledge on the number of adsorbed guest molecules in the pores of the material at operating conditions.
It is very hard to deduce this particular information from experimental data. This is especially true for applications taking place at higher temperatures. Therefore, there is a very strong incentive to compute the adsorption properties by means of molecular simulations. Furthermore, some of these nanoporous materials are very flexible, i.e. the volume and shape of the unit cell can change under influence of temperature, pressure or the interaction with the adsorbing species. A well-known example of such a material is the metal-organic framework MIL-53. This flexible behavior greatly influences the adsorption properties and can hence not be neglected in a computational investigation.
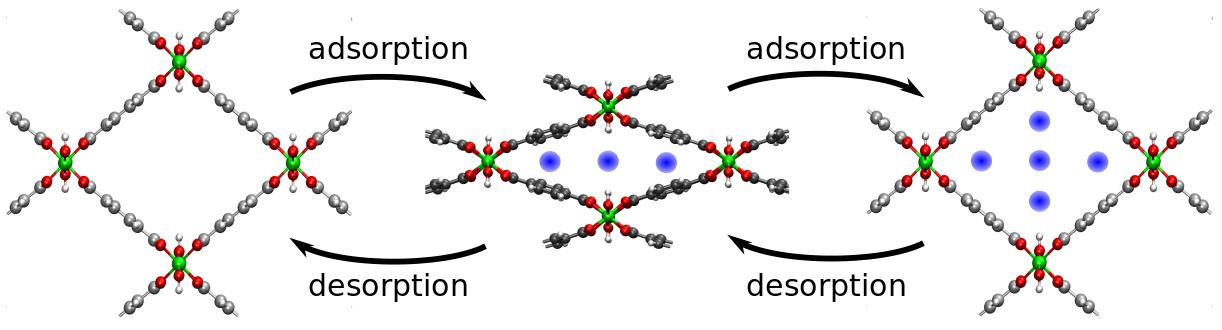
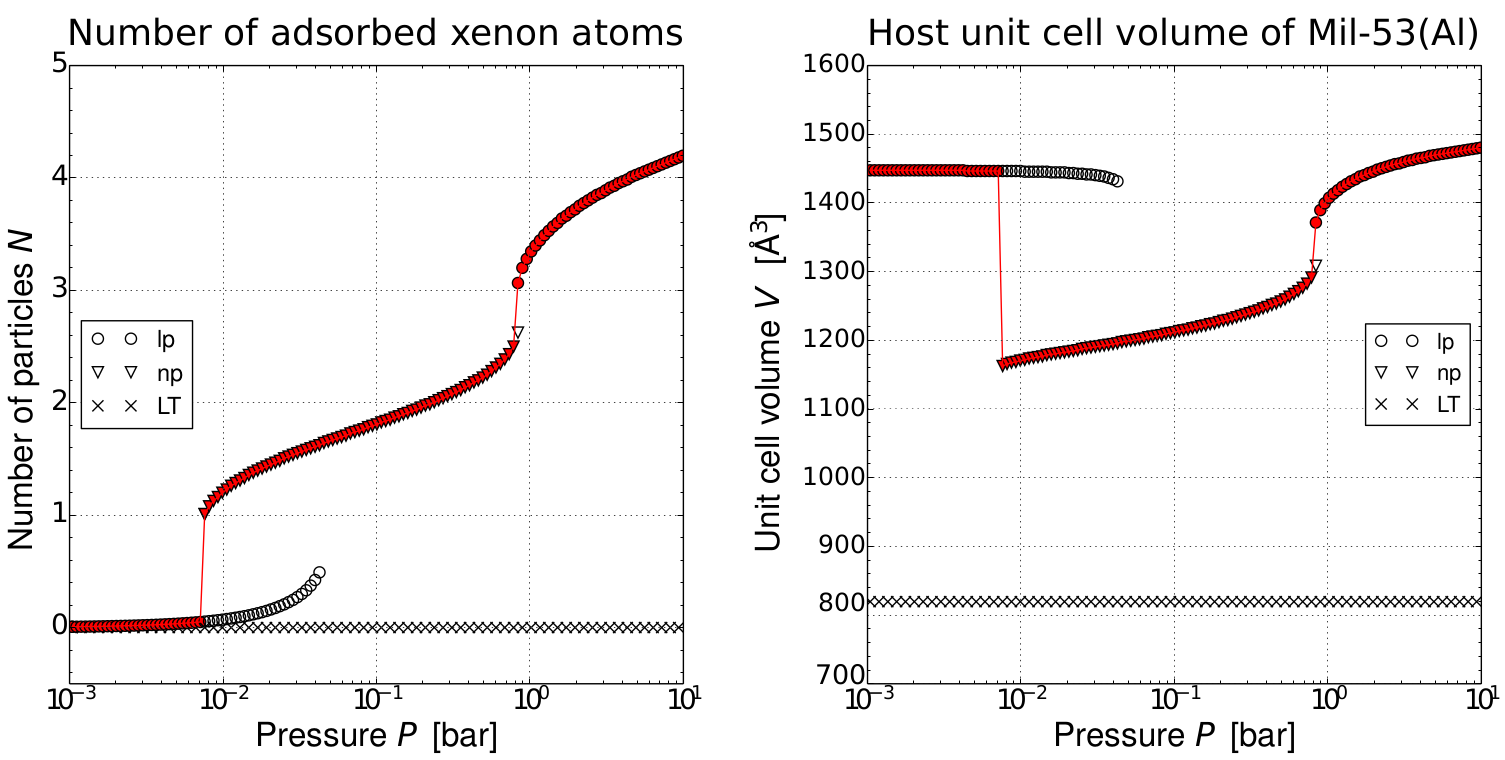
One procedure to obtain information on the loading of guest molecules in a nanoporous material is the construction of adsorption isotherms, i.e. the number of adsorbed species for a given vapor pressure. Current simulation methods mostly calculate these isotherms using a fixed volume and shape. At higher temperatures, this may be a drastic approximation. Another method to estimate the number of particles in the host material is the usage of a recently developed thermodynamic model at the Center for Molecular Modeling. This model starts from the free energy as function of fixed number of particles, fixed volume and fixed temperature, which is later translated to conditions of fixed pressure and chemical potential through the Legendre transform. The main approximation of this model is the use of a mean-field model to estimate the host-guest interaction. Although this mean field predicts adsorption isotherms that are in agreement with experiment, its validity has not yet been tested thoroughly.
To characterize the number of particles in the host material, one typically needs to describe phenomena at larger length and time scale, than what is feasible with quantum mechanical based methods. Therefore such simulations typically use force field based methods where the inter and intramolecular interactions of the nanoporous materials and guest species are described by a mathematical expression for the potential energy that is inspired by elastic springs connecting neighboring atoms. These force fields can be parameterized to mimic quantum mechanical input data. An automated protocol has been developed at the Center for Molecular Modeling to derive such force fields.
Goal
The goal of this thesis is to use so-called grand canonical Monte Carlo (GCMC) simulations to compute the adsorption isotherm and compare it with the application of the thermodynamic model. As such the GCMC results can be used to go beyond the mean-field approximation and improve the thermodynamic model.
At first instance, so-called grand canonical Monte Carlo (GCMC) simulations will be performed to compute the adsorption isotherm at fixed volume and shape. In such a simulation, the unit cell of the nanoporous host is kept fixed as well as the temperature and chemical potential. The number of guest molecules inside the pores, however, can fluctuate. These fluctuations are driven by an equilibrium with a reservoir at the given chemical potential and temperature. When the Monte Carlo simulation is equilibrated, the average number of particles inside the host during the entire simulation represents the equilibrium number of adsorbed particles at the given temperature, chemical potential and unit cell. By varying the chemical potential (at a fixed temperature), one can construct the adsorption isotherm. Furthermore, for flexible hosts, one can repeat the procedure for various unit cell configurations to investigate the influence of the framework flexibility.
In a second step, the results of the GCMC computations can be used to validate a thermodynamic model for adsorption in flexible MOFs developed at the CMM. This model starts by expanding the free energy in the NVT ensemble in three terms: the free energy of the empty host, the free energy of the guest molecules trapped inside the pores (without interacting with it) and the interaction of the guest molecules with the host. All three terms can be expressed analytically in terms of unit cell volume and number of particles. Finally, by means of the Legendre transformation from statistical physics, one can transform the free energy in the NVT ensemble to the thermodynamic potential in the μPT ensemble to predict the number of adsorbed species as a function of chemical potential at fixed temperature, i.e. the adsorption isotherm. The interaction term in the expansion is usually approximated in a mean-field context, which mainly implies two things: (1) the adsorption energy of N particles is equal to the N times the adsorption energy of 1 particle (2) the adsorption energy of a single particle inside the pores can be computed by keeping the atomic configuration of the host fixed. Both approximations have been proven to be adequate for adsorption of methane, carbon dioxide and xenon in MIL-53. However, the approximation has never been tested thoroughly yet. By comparing the results of the GCMC simulations with the thermodynamic model, the student will not only be able to express whether or not the model is valid, but the student will also be able to extend the mean-field approximation to improve the accuracy of the thermodynamic model.
During this topic, the student will expand his knowledge, insight and experience with thermodynamics, statistical physics and molecular simulation. Programming skills required for efficient processing of data will be transferred during the thesis if it is required.
Aspects
Physics: This subject requires a applied knowledge of thermodynamics and statistical physics / Engineering: Molecular simulations are applied to investigate the adsorption applications of nanoporous materials


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELLING, NANOKeywordsAdsorption, Thermodynamics, grand canonical monte carlo, Nanoporous materials
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck