No more randomness: computational NMR to spatially resolve the aluminum distribution in zeolites
No more randomness: computational NMR to spatially resolve the aluminum distribution in zeolites
Promotor(en): V. Van Speybroeck /20SPEC03 / SpectroscopyZeolites are crystalline aluminosilicates that are used for a broad range of industrial applications such as gas separation and capture and as highly efficient catalysts. Their structure consists of tetrahedrally coordinated SiO4/AlO4 units connected in such a way that the framework constitutes a network of pores and channels. For each aluminum tetrahedron, there must be a countercharge present to maintain charge neutrality of the framework. This countercharge will act as an acid site and is introduced in the zeolite either through extraframework species or as a proton bound to one of the four neighboring oxygen atoms. Owing to their nanoporosity, adsorbates can be present in the structure and use the available acid sites to accelerate chemical processes such as the methanol-to-olefin reaction. The actual reactions taking place heavily depend on the structure and position of the acid sites in the zeolite. To increase the efficiency of industrial processes and to be able to tune the aluminum distribution, it is vital to accurately characterize the precise nature and distribution of the aluminum species.
To explain the relationship between structure and corresponding reactivity, zeolites can be investigated through operando spectroscopy. Nuclear magnetic resonance (NMR) spectroscopy is a technique that is particularly useful to resolve complex structures as it is extremely sensitive to the local environment of the isotope under investigation. Nuclear spins in a strong magnetic field are perturbed through a small oscillating field, and their response causes a resonance of which the frequency is strongly dependent on the electronic structure surrounding the nucleus. By analysing the NMR spectrum, structures can be resolved that are too complex for techniques insensitive to short-range interactions such as X-ray diffraction [1]. Despite important methodological developments (e.g. magic-angle spinning) and technological advances (e.g. stronger magnetic fields, faster spinning rates), there is a growing need for computational techniques to improve the interpretation of experiment. Multiple algorithms have been developed in the course of the last two decades, enabling the simulation of complex materials such as zeolites and the computation of the NMR properties via quantum chemical calculations. In this regard, the locations of distinct acid sites can be and have been characterized through NMR spectroscopy [2].
Although the calculation of NMR properties has become a straightforward procedure, it remains often difficult to match theoretical predictions with experimental observations. In order to learn something from theory, it is vital to simulate the zeolite structure under the right physical conditions. This is challenging because, for a given number of aluminum tetrahedra, there exist a vast amount of possible acid site configurations, which all give rise to slightly different NMR properties but lead to distinctly different catalytic efficiency of the zeolite. Furthermore, experimental NMR spectra of zeolites are usually recorded while water and/or other adsorbates are still present in the pores. This can also have major implications for the spectroscopic results [3,4]. Finally, aluminum may not only be present inside the framework itself, but it may also exist as an extraframework species. However, the nature of possible extraframework aluminum (EFAL) species is not yet clear, as they might be present as mononuclear or multinuclear structures.
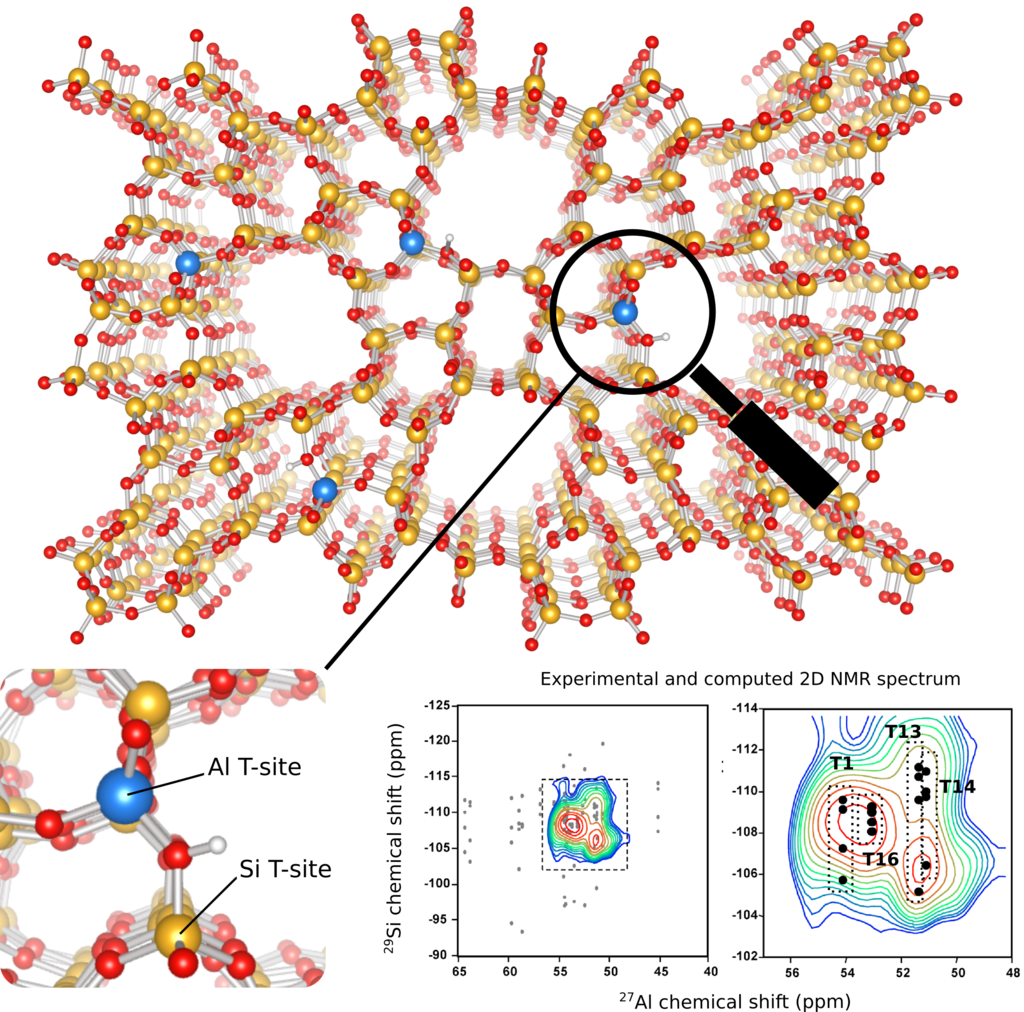
Figure 1: Example of a zeolite framework with aluminum (blue) and silicon (yellow) atoms. By combining NMR spectroscopy with quantum chemical calculations, the Al distribution in a given sample can be assigned to specific crystallographic T-sites. (data adapted from ref. [2])
Doelstelling:
This thesis aims to apply state-of-the-art computational techniques to investigate the NMR properties for a range of important zeolite frameworks under different conditions. A first step towards the in spatio characterization of acid site configurations is the reduction of the computational cost due to the many possible aluminum distributions. While the number of configurations increases roughly exponentially with the number of acid sites per unit cell, the amount of relevant configurations lies much lower. Many equivalent configurations can be eliminated based on symmetry considerations. Possible trends in the calculated NMR parameters can then be investigated based on a relatively small subset of relevant structures.
A second aspect in this thesis will be the role of water on the resulting NMR spectra. While it is obvious that NMR parameters will be affected (sensitivity to the local environment!), both the nature of the interactions and the extent of their influence are unknown. At first instance, static calculations will be performed, in which temperature effects are ignored. However, as water can be very mobile in the zeolite pores, it will also be necessary to apply computationally heavier dynamic methods that allow to simulate the material at a certain temperature. Combining both static and dynamic approaches, the behaviour of water in the pores can be investigated and the implications for the NMR properties can be assessed. It is known that the proton can be solvated to form charged hydronium complexes. In order to judge whether this has implications for reaction mechanisms (is the proton still available for catalysis?), it is important to know at which framework loading (number of water molecules per pore) this solvation can take place. The same questions can be asked about other adsorbates that can be hosted by the zeolite.
The last part of this thesis will focus on the nature of EFAL species in zeolites. EFAL species can be formed as mononuclear or multinuclear structures. Knowledge of the precise structure is important, as this can largely affect the catalytic properties. For example, very large EFAL species might hinder the diffusion of other guest molecules, hence reducing the catalytic activity. The NMR properties will be studied via dynamic approaches to closely mimic the exact physical conditions.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsZeolites, NMR spectroscopy, Nanoporous materials, Density functional theory, extraframework species, nanoscale modelingReferences
[1] D. H. Brouwer et al., JACS 127, 10365-10370 (2005)
[2] E. Dib et al., J. Phys. Chem. Lett. 9, 19-24 (2018)
[3] R. E. Fletcher et al., Chem. Sci. 8, 7483-7491 (2017)
[4] C. J. Heard et al., Chem. Sci. 10, 5705-5711 (2019)
Contact
Veronique Van Speybroeck