A molecular dynamics approach to electron-phonon coupling in photovoltaic perovskites
A molecular dynamics approach to electron-phonon coupling in photovoltaic perovskites
Promotor(en): V. Van Speybroeck, K. Lejaeghere /19SPEC02 / SpectroscopyPerovskites are well-known minerals with an ABX3 stoichiometry. Named after the Russian mineralogist Lev Perovski, their orthorhombic crystal structure has already been identified in the early 20th century. Because of their strong coupling between crystal symmetry and directional material properties (e.g. dipole moment), perovskites have now attracted renewed interest: they exhibit exciting new properties such as ferro-electricity (spontaneous polarization) and ferro-elasticity (spontaneous strain).
One particular class of perovskites, hybrid organic-inorganic perovskites (HOIPs), are under world-wide investigation for an entirely different reason: they can be applied for photovoltaic (PV) purposes. HOIPs contain both inorganic and organic components. The most famous HOIP currently used is methylammonium (MA) lead iodide (MAPbI3), where the organic methylammonium cation is encaged in a negatively charged lead iodide framework. Since its first consideration as a PV material in 2009, the efficiency of perovskite-based solar cells has gone up from 3.8 % to 22.1 %. This strong growth over only a few years makes many researchers believe that perovskites have not yet reached their limits. Their easy synthesis, low cost and tunable band gap make them promising competitors for traditional PV materials.
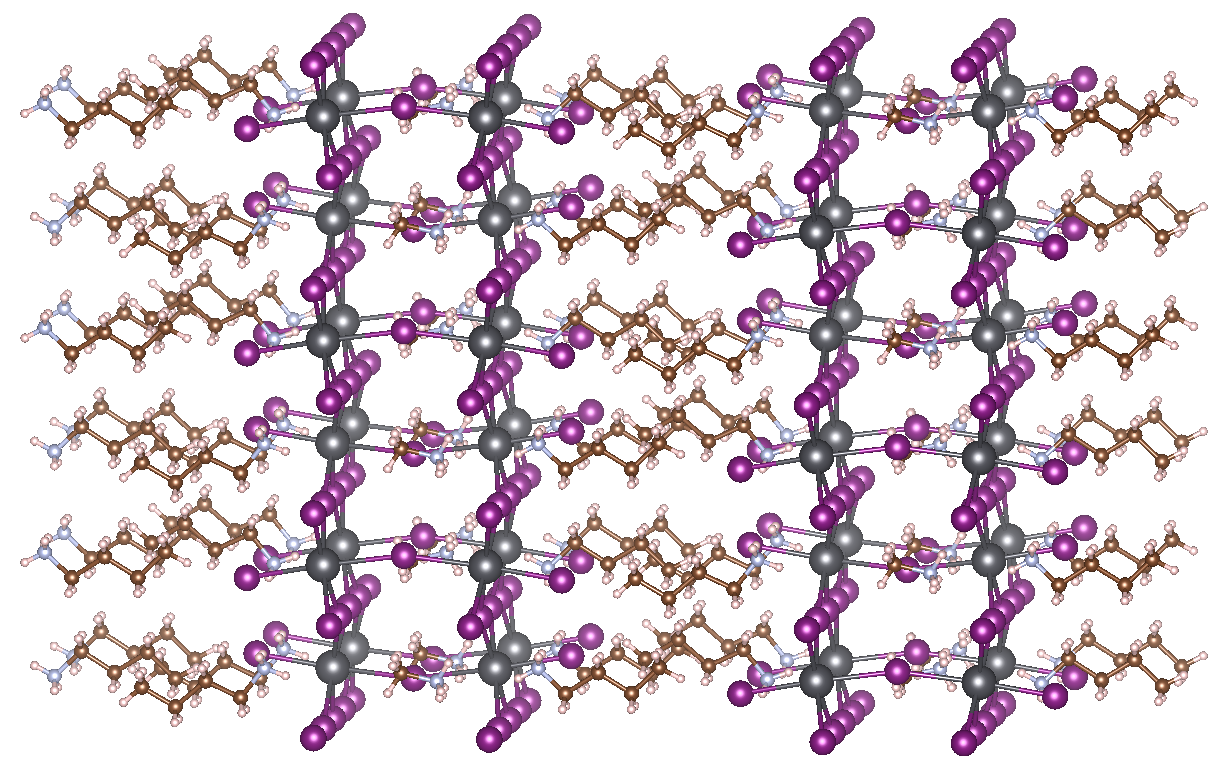
Contrary to more common solar cell materials, perovskites possess a huge structural variability. This is because the central cation can move around quite easily, which distorts the local geometry of the anionic cages. This structural freedom affects the band structure of the perovskite and hence the efficiency of the PV device. Electron-phonon coupling gives rise to a significant broadening of photoluminescent emission lines, for example, but also influences charge transport properties. Some preliminary research has already been performed to shed light on predominant effects in lead halide HOIPs, but it is still not entirely clear which vibrational modes influence which electronic energy levels. In addition, researchers are now increasingly turning to so-called Ruddlesden-Popper perovskites for their improved humidity resistance. These materials consist of 2D layers of traditional perovskite material, interleaved with cations. In the case of 2D HOIPs, these spacer cations are organic as well, with the most prominent example being MAPbI3 interleaved with n-butylammonium (BA) (see figure). The interaction between vibrational and electronic degrees of freedom in these materials is still largely unknown.
Goal
The goal of this thesis is to perform an in-depth study of the coupling between vibrational and electronic degrees of freedom in 3D and 2D HOIPs using density-functional theory (DFT). DFT is a quantum physical method that allows describing the electron behaviour in realistic materials at an acceptable cost. You will be able to employ both the CP2K and the VASP package, for both of which there is extensive expertise at the Center for Molecular Modeling. To assess the vibrational behaviour of hybrid perovskites, you will use molecular dynamics (MD). This approach allows simulating a material as a function of time by integrating Newton’s equations of motion and taking into account temperature and pressure. A Fourier transform of the time-dependent atomic movements then identifies the predominant phonon modes. These isolated phonon modes can be attributed to particular bond environments and therefore constitute a fingerprint of the considered material. The dynamic electron behaviour can be investigated via electronic-structure calculations at given snapshots of the MD simulation. This gives you temperature-dependent band gaps and charge carrier mobilities. The most important frequencies in these fluctuations can again be determined from a Fourier transform. The agreement between the phonon frequencies and the frequencies underlying the electronic variations then indicates where electron-phonon coupling is substantial. You can calculate how these phonon modes look like by means of a principal component analysis. Your thesis will consist of two major parts. First, you will need to establish an efficient methodology to perform the intended electron-phonon comparison. Important aspects are the used level of theory and the settings of the molecular dynamics run. You will therefore start investigating 3D perovskites, which allows you to compare to experimental and theoretical literature and extend insights to e.g. tin halides, which are less toxic than lead-containing HOIPs. In a second part, you will then be able to use your experience to tackle the challenging case of 2D HOIPs. Based on your results, you will be able to tune parts of the structure to adapt the observed behaviour.
Context for Engineering Physics students
For students in Engineering Physics, this thesis subject is closely related to the following clusters of elective courses: MODELLING, MATERIALS, NANO, ELECTRONICS and PHOTONICS. Physics aspects: use of and insight in quantum mechanical models for materials modelling; engineering aspects: application to materials properties and semiconductor engineering

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELLING, MATERIALS, NANO, ELECTRONICS, PHOTONICSKeywordshybrid inorganic-organic perovskite, density-functional theory, Molecular dynamics, electron-phonon couplingRecommended coursesSimulations and Modeling for the Nanoscale, Computational Materials PhysicsReferences
A. Mattoni et al., ‘Modeling hybrid perovskites by molecular dynamics’, J. Phys.: Condens. Matter 29, 043001 (2017). http://dx.doi.org/10.1039/C4TA05033A.
Y. Chen et al., ‘2D Ruddlesden-Popper Perovskites for Optoelectronics’, Adv. Mater. 30, 1703487 (2017). http://dx.doi.org/10.1002/adma.201703487.
A. Van Yperen-De Deyne et al., ‘Exploring the Vibrational Fingerprint of the Electronic Excitation Energy via Molecular Dynamics’, J. Chem. Phys. 140, 134105 (2014). http://dx.doi.org/10.1063/1.4869937.
Contact
Veronique Van Speybroeck