Modelling physisorption and swelling by alcohols in layered nanoporous frameworks
Modelling physisorption and swelling by alcohols in layered nanoporous frameworks
Promotor(en): V. Van Speybroeck, K. Lejaeghere /18NANO15 / Nanoporous materialsIn recent years, crystalline nanoporous materials have attracted much interest owing to their numerous applications, such as gas/liquid storage and separation, catalysis and drug delivery. One such class of materials are covalent organic frameworks (COFs), which can be thought of as the condensation of one or several building blocks, resulting in an ordered structure of organic moieties connected with strong covalent bonds. This modular approach is referred to as reticular synthesis and endows COFs with a huge tunability. COFs moreover have the additional advantage that they do not contain any metals. This not only makes them lighter than traditional porous solids, but also yields catalysts that are more environmentally friendly.
Reticular synthesis of COFs usually results in either 3D or 2D materials, where the latter consist of stacked, mostly planar sheets. Because of the absence of bonding interactions between the layers of 2D COFs, these materials inherently possess a large conformational freedom. That freedom is expressed by both the layer stacking and their interlayer distance, and results from a subtle interplay between the non-bonding interactions, such as London dispersion. The figure below shows the example of a 2D COF, which can display a different stacking order and distance depending on the make-up of the framework, temperature and pressure, and the strength of the nonbonding interactions.
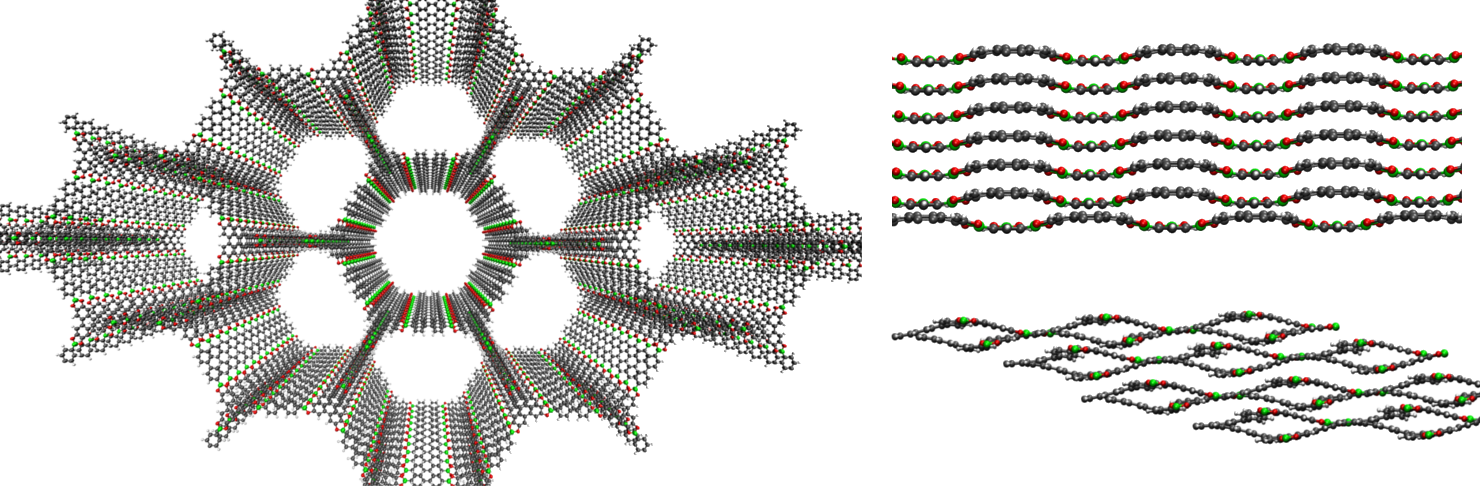
Covalent triazine-based framework (CTFs) are a particularly promising type of 2D COFs that can be used for the selective oxidation of alcohols into aldehydes and ketones. However, experiments show that when loaded with alcohols, the CTFs behave unexpectedly. For example, it appears that the interlayer distance increases dramatically when introducing methanol into the 2D COF, while this effect is not observed upon water adsorption. In addition, the position of the adsorbents as well as their interaction strength with the CTF are not well understood. In general the question arises to what extent the stacking order, porosity and interlayer distance is influenced as a result of introducing different guest molecules between the layers.
Goal
The goal of this thesis is to investigate the interplay between adsorbed organic molecules and CTFs. You will use both density-functional theory (DFT) and classical force fields (FF). DFT is a quantum physical method that allows describing the full electron behaviour, while FF approaches reduce the computational complexity by condensing electrons and nuclei into atomic entities connected through classical bonding and nonbonding interactions. For both DFT and FF calculations, you can use available software packages for which there is extensive expertise at the Center for Molecular Modeling. An important aspect to consider, will be how to describe the nonbonding interactions, both when using DFT and when using FF.
During your thesis, you will screen a number of different adsorbent molecules to see how this affects the adsorption into a CTF material. Both aromatic and aliphatic molecules will be considered, and their size will be varied. In addition, the composition of the CTF itself can be changed as well, following the idea of reticular synthesis. By changing the linker length, width and aromaticity, the adsorption of the system may differ significantly.
You will perform two types of simulations. On the one hand, much information comes from static calculations, in which the properties of the system are computed at fixed geometries. This will allow you to say something about the adsorption strength, the most favourable position of the adsorbent and the layer stacking in the CTF. Dynamic simulations, on the other hand, provide more insight into the degrees of freedom accessible by temperature or pressure. In addition, you can use grand canonical Monte Carlo (GCMC) to dynamically change the number of adsorbed particles, which yields the optimal number of adsorbed molecules in the system as a function of temperature. Using these techniques, you will be able to elucidate the thus far poorly understood swelling of CTFs under alcohol adsorption.
You will be actively coached to acquaint yourself with the required simulation techniques early in the thesis year. In addition, this work is inspired by experimental collaborators at the UGent Center for Ordered Materials, Organometallics and Catalysis (COMOC). Your work will take place in close contact with these partners.
Context for Engineering Physics students
Physics: use of and insight in quantum mechanical models for materials modelling
Engineering: application to materials properties

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELING, MATERIALS, NANOKeywordsCovalent organic frameworks, Two-dimensional material, Adsorption, density-functional theory, Molecular dynamicsRecommended coursesSimulations and Modeling for the NanoscaleReferences
P. Kuhn et al., ‘Porous, Covalent Triazine-Based Frameworks Prepared by Ionothermal Synthesis’, Angew. Chem. Int. Ed. 47, 3450-3453 (2008). http://dx.doi.org/10.1002/anie.200705710
B. Lukose et al., ‘On the reticular construction concept of covalent organic frameworks’, Beilstein J. Nanotech. 1, 60-70 (2010). http://dx.doi.org/10.3762/bjnano.1.8
Contact
Veronique Van Speybroeck