Modeling the adsorption of guest molecules in flexible metal-organic frameworks via hybrid MC/MD schemes
Modeling the adsorption of guest molecules in flexible metal-organic frameworks via hybrid MC/MD schemes
Promotor(en): V. Van Speybroeck, T. Verstraelen /16MODEV12 / Model and software developmentIn tackling the climate change problem, the reduction of greenhouse gas emissions plays a major role. These emissions, for which CO2 and CH4 are the main perpetrators, can be efficiently reduced by adsorbing these harmful gas molecules inside a host material (see figure 1), after which these gases can either be separated or sequestered. Several potential host materials are being studied, but it is safe to say nanoporous materials, and within this class, metal-organic frameworks (MOFs), are viable materials to fulfill this aim. MOFs are scaffold-like structures obtained by linking inorganic bricks by organic ligands, obtaining a nanoporous structure (see figure 1). The large internal surface of these materials makes them excellent candidates for the aforementioned gas storage applications, since they optimize the affinity between the guest and the host material. Moreover, a lot of these MOFs exhibit linker flexibility, so that pore sizes can vary, or different porous channels can merge, hence clearing the way for guest adsorption. Consequently, to computationally characterize a MOF’s potential for guest adsorption, it is paramount to incorporate this flexibility in an adequate way. Most contemporary studies neglect this flexibility by sampling in the grand canonical ensemble with a rigid framework, which may be a severe approximation [1].
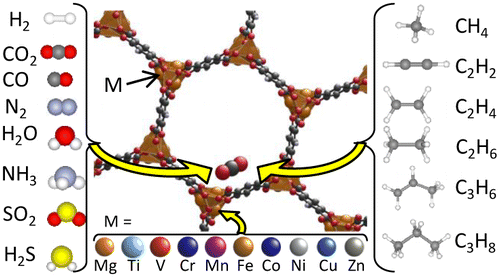
Goal
The aim of this thesis is to overcome the rigid framework approximation by constructing an advanced molecular simulation scheme. This hybrid scheme will be composed of two contributions: a molecular dynamics (MD) contribution in which the Newtonian equations of motion are integrated, and a Monte Carlo (MC) contribution which can be used to insert or remove guest molecules. As such, both the ‘true’ dynamics, and hence the flexibility, of the MOF will be described (in the MD part), while allowing the number of guest molecules to change (in the MC part), something that is impossible in a pure MD simulation. To develop this scheme, ideas can be borrowed from MD and MC codes that were recently implemented in our in-house Pythonic molecular simulations engine, Yaff. However, the MC/MD hybrid scheme will probably pose additional hurdles which needs to be tackled by the student.
While the student is encouraged to envisage its own hybrid schemes once familiar with the topic, two existing schemes are already defined (see figure 2). In a first scheme, an MD simulation of an empty MOF is run for a sufficiently long time, and several snapshots of the MOF are taken during this run. These different snapshots are expected to be representative for the flexibility of the host material. Afterwards, a separate MC simulation is started for each snapshot. During these MC simulations, we try to add (or remove) guest molecules to the host material, something that will only succeed when the guest-host interactions are favorable. When averaging over all these snapshots, the average amount of guest adsorption in the MOF can be obtained [2]. In a second scheme, a short MD simulation is initiated, after which we try to add or remove a guest molecule via an MC step. After this MC step, whether successful or not, we again perform an MD simulation, possibly with a different number of guest molecules. As such, we will obtain a sequence of alternating MD and MC steps, so that guest molecules can be removed or added during the hybrid simulation. The average amount of guest molecules is then again obtained by averaging over the whole MD/MC run [3]. While this second scheme represents more the ‘true’ phenomenon of guest adsorption in a MOF, special attention should be paid to ensure detailed balance during the MD run–something the student will become familiar with early in the thesis.
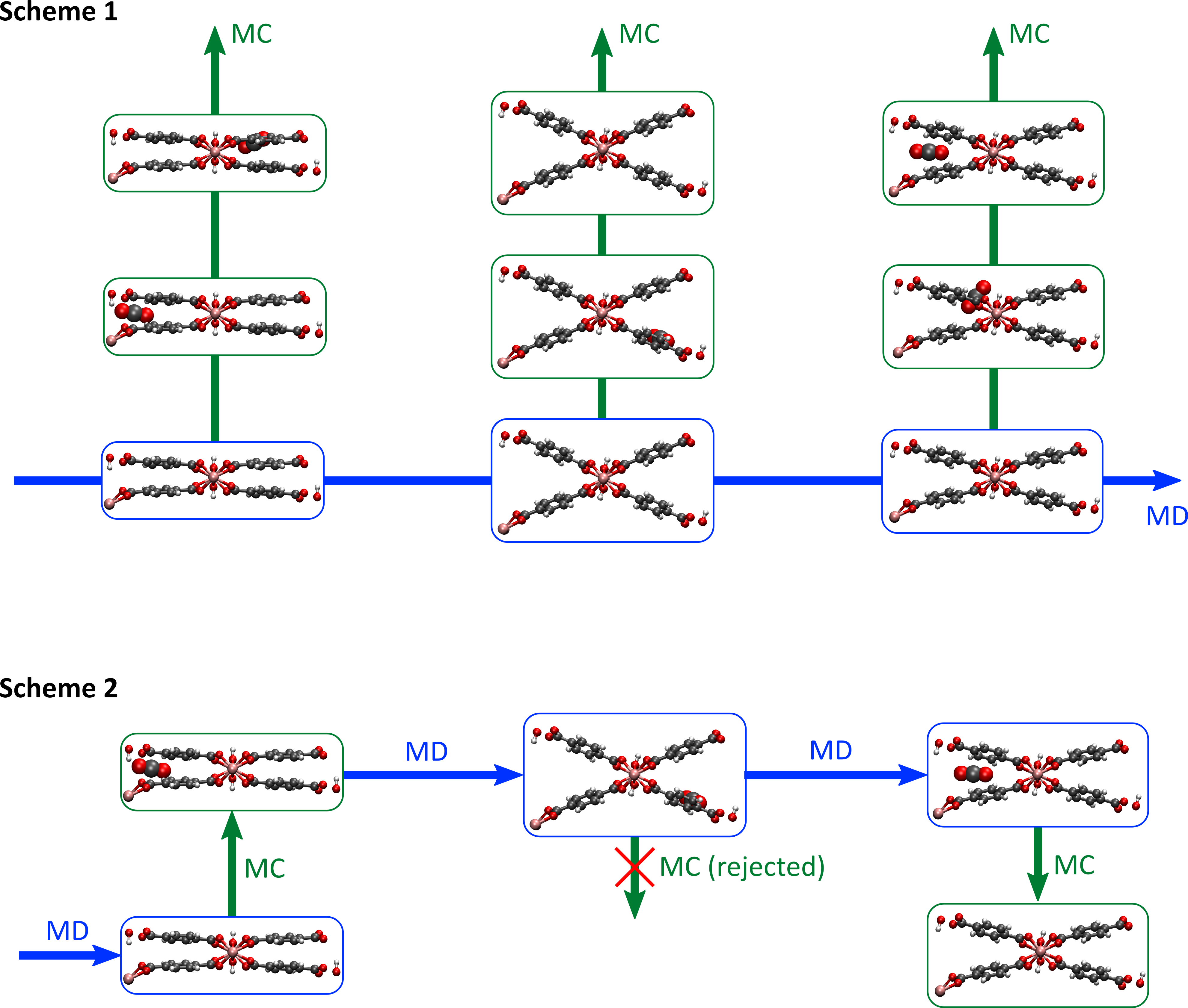
The goal of this thesis subject is hence threefold. First, the student needs to get acquainted with the fundaments of Monte Carlo and molecular dynamics techniques, which form the basis for any molecular simulation. Second, these two techniques need to be combined and implemented following the two schemes outlined above, potentially extended to include a scheme developed by the student. Hence, it is expected the student also looks forward to implement their own software code to extend the existing molecular engine. Lastly, using this new methodology, the adsorption of for instance CO2 and CH4 in several MOFs will be assessed to give an indication of the best-performing MOFs in class for the reduction of greenhouse gases.
Aspects
Physics aspect: The interaction between guest molecules and the host material will play a dominant role in this thesis subject. Moreover, the student will explore the fundaments of molecular simulations.
Engineering aspect: The aim of this thesis is to obtain well-performing materials for the adsorption of greenhouse gases, contributing to the quest to curb climate change.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELING, NANOKeywordsImplementation, guest adsorption, Force fields, Molecular dynamics, Monte Carlo, FlexibilityReferences
[1] F.-X. Coudert en A. H. Fuchs, „Computational characterization and prediction of metal-organic framework properties,” Coord. Chem. Rev., vol. 307, nr. 2, pp. 211-236, 2016.
[2] J. A. Gee en D. S. Sholl, „Effect of framework flexibility on C8 aromatic adsorption at high loadings in metal-organic frameworks,” J. Phys. Chem. C, vol. 120, nr. 1, pp. 370-376, 2016.
[3] D. Dubbeldam, S. Calero, D. E. Ellis en R. Q. Snurr, „RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials,” Mol. Simulat., vol. 42, nr. 2, pp. 81-101, 2016.
Contact
Veronique Van Speybroeck
Toon Verstraelen