A mindless exploration of the free-energy surface: accelerated Molecular Dynamics and Nested Sampling
A mindless exploration of the free-energy surface: accelerated Molecular Dynamics and Nested Sampling
Promotor(en): T. Verstraelen, A. R. Mehdipour /28305 / Model and software developmentBackground and problem
Molecular Dynamics (MD) simulations integrate Newton’s equations of motion of atomistic models. Forces acting on the nuclei are computed at each time step to propagate the molecular structure through time. While the resulting MD trajectories can elucidate some macroscopic features of a molecular model, a standard MD simulation is generally too inefficient to characterize experimentally relevant properties, such as an overview of all thermodynamically relevant states, and the transitions between them. MD simulations are often enhanced, using modified algorithms, to improve the efficiency with which they explore the potential-energy surface. Only with specialized hardware, such as GPUs or the ANTON computers, plain MD is sometimes still effective. Obviously, also on such advanced hardware, more efficient sampling algorithms are still of interest. Most forms of enhanced MD require some user-specified bias in the potential-energy surface, e.g., one may artificially destabilize a state and drive the transition to another relevant meta-stable state. The downside of this user input is that the exploration is limited by the creativity of the operator. Fully mindless enhanced MD is uncommon, yet highly relevant to discover unexpected results.
This MSc thesis focuses on the development of an enhanced MD method. Short peptides will be used as test cases, because (a) peptide MD on GPU hardware is readily available, (b) short peptides are well understood and (c) they are still biologically relevant, e.g., as biopharmaceuticals.[1] That being said, no prior background in biochemistry is needed for this topic. Instead, a solid understanding of statistical physics and computational skills are the main prerequisites. Once developed, the methodology from this MSc thesis should also be easily applicable to other molecular systems.
Nested Sampling (NS, see Figure 1) is a recent development in statistics (proposed less than 20 years ago)[2] and has only occasionally been applied to molecular simulations, including short peptides.[3] From a physics perspective, NS is an athermal method, in which the temperature is fixed only during post-processing. The original NS method has some practical limitations and is not trivially applicable to molecular systems. These limitations were addressed in more recent literature [4-6] and a common strategy in those articles is to use MD as a building block in the NS algorithm.
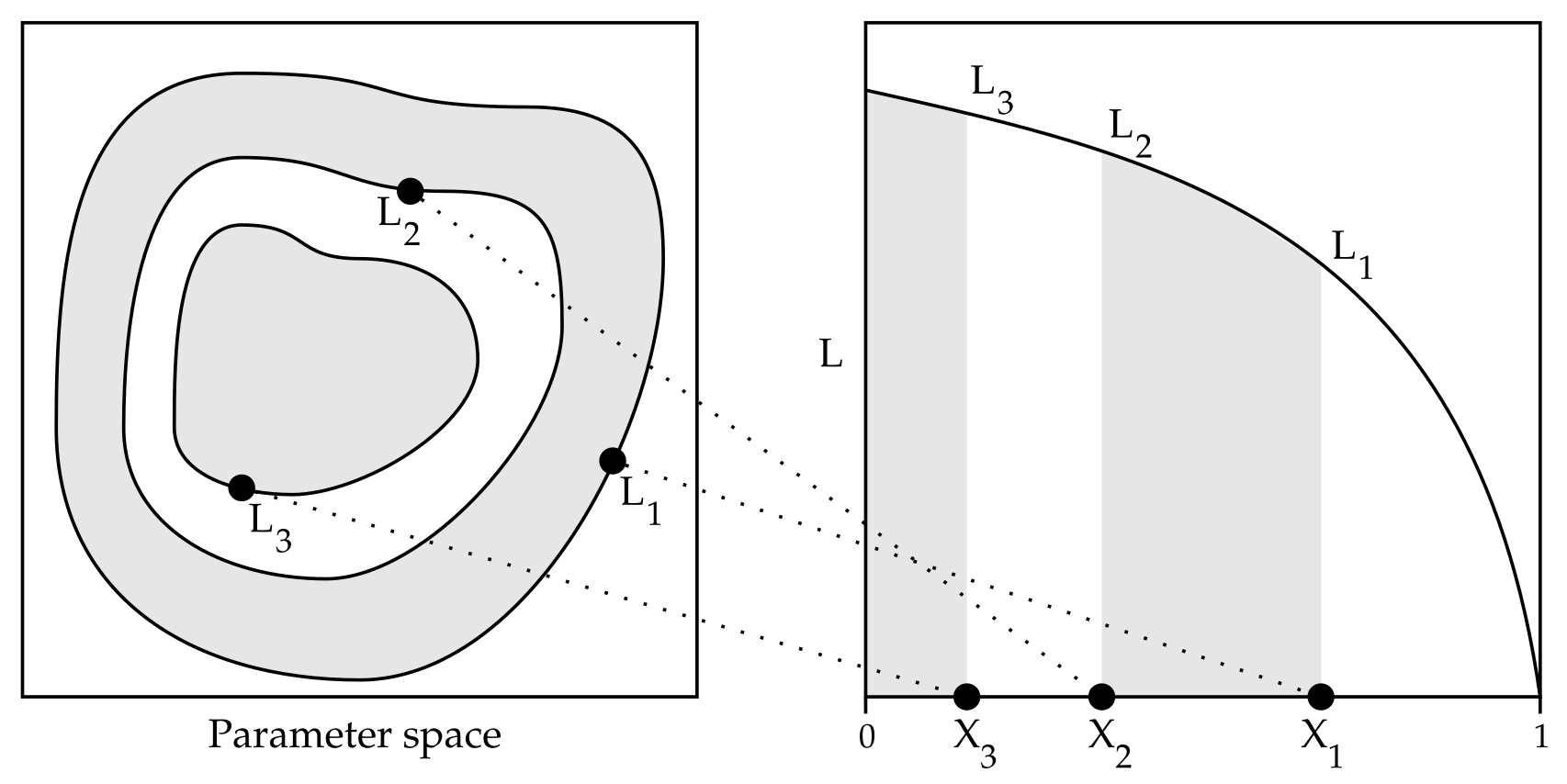
Figure 1: Illustration of the basic principle of Nested Sampling (NS) proposed by Skilling (2006).
Goal
The goal of this MSc thesis is to combine the so-called accelerated MD (aMD, see Figure 2) method [7] with nested sampling (NS). Even though these two methods are distinct and were developed independently, they have some common features, which can be exploited. The first step in the thesis is to work out a theoretical framework and an algorithm combining aMD and NS. The algorithm will be implemented in an open-source Python module, using OpenMM for the MD simulations. (OpenMM is a highly efficient MD library for biomolecules with an intuitive Python interface.) Finally, the implementation will be tested on a series of short peptides with known conformational preferences.[8] The final objective is to recover these preferences without any user-specified bias in the simulations.
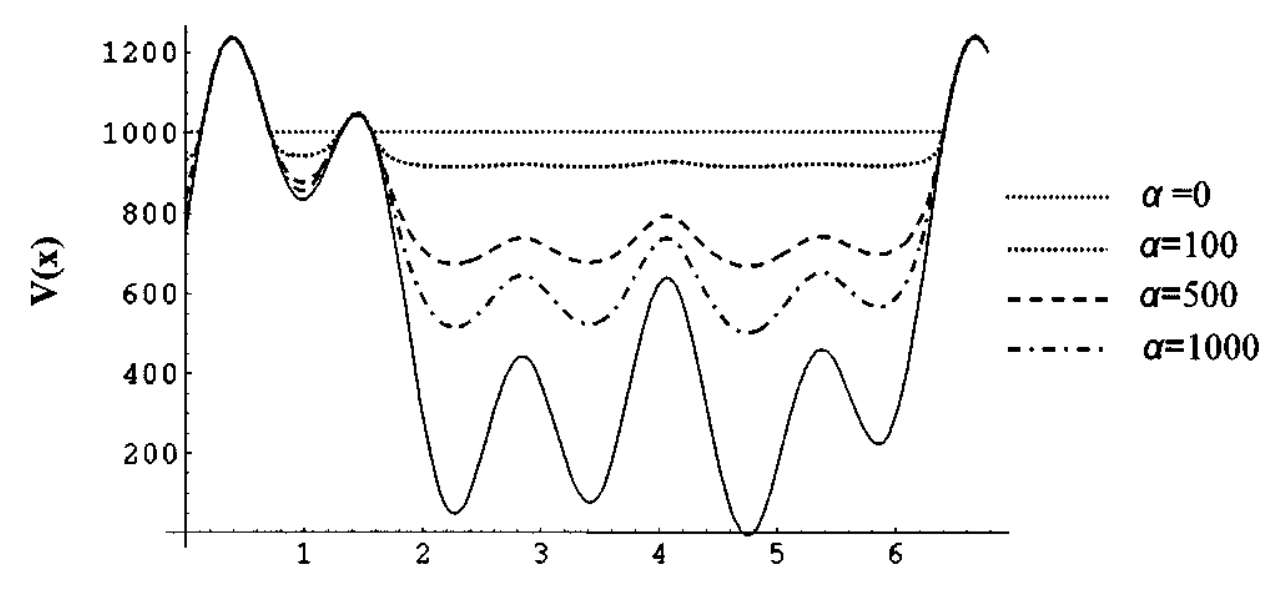
Figure 2: Illustration of the modified potential energy surface in accelerated Molecular Dynamics (aMD), as proposed by Hamelberg. (2004)
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]Keywordsaccelerated Molecular Dynamics, Nested Sampling, Free EnergiesReferences
[1] Apostolopoulos et al., Molecules, 26, 430 (2021) https://doi.org/10.3390/molecules26020430
[2] Skilling, Bayesian anal., 1, 833 (2006) https://doi.org/10.1214/06-BA127
[3] Burkoff et al., Computer Physics Communications, 201, 8 (2016) https://doi.org/10.1016/j.cpc.2015.12.005
[4] Nielsen, J. Chem. Phys., 139, 124104 (2013) https://doi.org/10.1063/1.4821761
[5] Martiniani 2014, Phys. Rev. X, 4, 031034 (2014) https://doi.org/10.1103/PhysRevX.4.031034
[6] Griffiths et al., J. of Chem. Phys., 15, 6865 (2019) https://doi.org/10.1021/acs.jctc.9b00567
[7] Hamelberg et al., J. Chem. Phys., 120, 11919 (2004) https://doi.org/10.1063/1.1755656
[8] Voelz et al., PLoS Comput Biol., 5, e1000281 (2009) https://doi.org/10.1371/journal.pcbi.1000281
Contact
Ahmad Reza Mehdipour
Toon Verstraelen