Mechanistic study on the early stages of the MTO process using advanced molecular simulations
Mechanistic study on the early stages of the MTO process using advanced molecular simulations
Promotor(en): V. Van Speybroeck /17NANO01 / Nanoporous materialsDue to the growing relevance of renewable resources, interest in the methanol-to-olefin (MTO) process has grown since it is one of the most prominent technologies to bypass crude oil in light olefin production [1]. This methanol conversion process is catalyzed by zeolite or zeotype catalysts. In particular, the chabazite-structured H-SAPO-34 is of industrial interest due to its high light olefin selectivity. Intensive research to elucidate the MTO reaction mechanism led to the general acceptance of the hydrocarbon pool (HP) mechanism, in which an organic species occluded in the zeolite framework acts as a co-catalyst.
Although the HP mechanism is generally accepted to govern the MTO process during the active period, the formation of the first carbon-carbon (C-C) bond still remains a puzzling question and thus an active research field. Many direct C-C formation mechanisms have been disregarded due to very high energy barriers that cannot be overcome at typical MTO conditions.[2] Some studies even assume that the presence of traces of impurities in the methanol feed leads to the first C-C bond formation [3]. Nevertheless, recent findings provide evidence for the crucial role of oxygen containing intermediates such as aldehydes during the formation of the initial C-C bound.[4, 5] In a recent experimental study, reaction mechanisms are proposed based on experimentally observed intermediates, i.e. surface methoxy species, surface-trapped formate and acetate species, methyl acetate and dimethoxymethane. This reaction mechanism for the first C-C coupling is summarized in Figure 1.
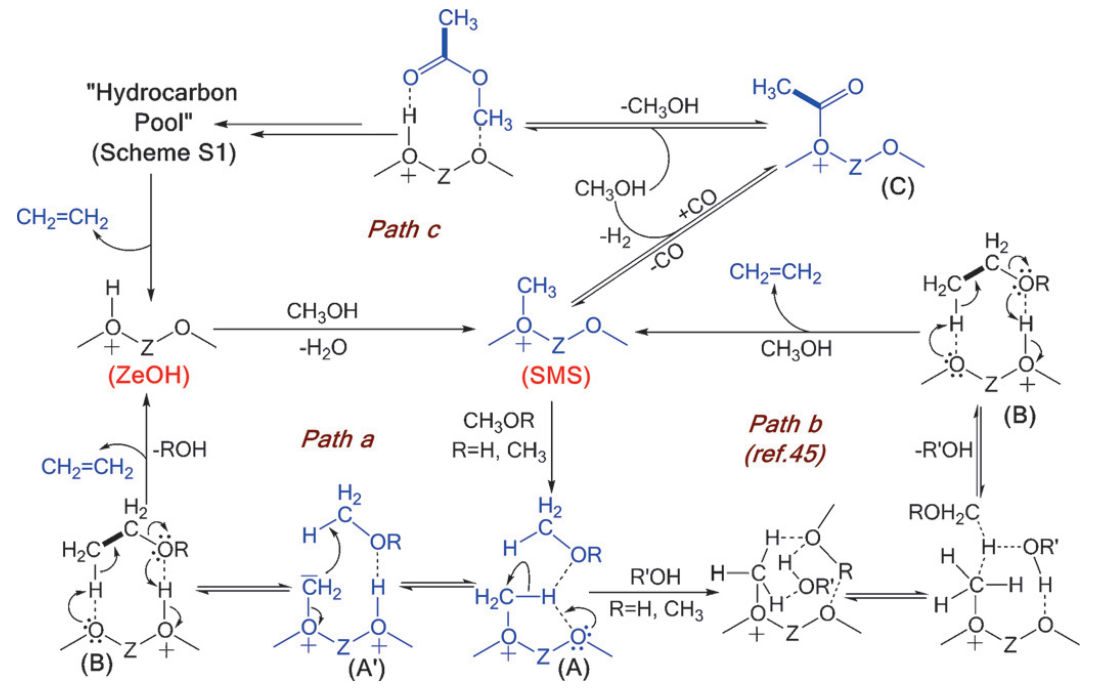
Figure 1: Schematic representation of possible reaction mechanisms for the formation of the first C-C bond in the MTO process.[4]Goal
The objective of this master thesis is to find theoretical evidence for the experimentally proposed C-C coupling reaction mechanisms in H-SAPO-34 at realistic MTO reaction conditions using advanced molecular modeling techniques. To this end first principle chemical kinetics will be calculated for the various elementary reactions. These simulations require the application of a variety of molecular simulation techniques, which rely on first principle static and molecular dynamics simulations. Further, the student can build a micro-kinetic model to assess the influence of operating conditions (temperature and pressure) on the formation of the first ethene molecules during the early stages of the MTO process.
The Center for Molecular Modeling has built up vast expertise in these advanced simulation techniques and collaborates on the subject with leading experimental and theoretical partners. The student will be actively coached to get acquainted with the plethora of techniques needed to tackle the proposed problem. It is the intention to involve the student actively in the work discussions with our collaborators. The CMM has access to sufficient computational resources to execute this research project. The proposed topic is challenging and requires technical skills, creativity and chemical insight.

- Study programmeMaster of Science in Chemical Engineering [EMCHEM]KeywordsHeterogeneous Catalysis, Chemical kinetics, Computational applicationsReferences
[1] K. Hemelsoet, J. Van der Mynsbrugge, K. De Wispelaere, M. Waroquier, V. Van Speybroeck, Unraveling Reaction Mechanisms governing Methanol-To-Olefins Catalysis Combining Theory with Experiment, ChemPhysChem, 14 (2013) 1526-1545.
[2] D. Lesthaeghe, V. Van Speybroeck, G.B. Marin, M. Waroquier, Understanding the Failure of Direct C-C Coupling in the Zeolite-Catalyzed Methanol-to-Olefin Process, Angew. Chem. Int. Ed., 45 (2006) 1714-1719.
[3] W.G. Song, D.M. Marcus, H. Fu, J.O. Ehresmann, J.F. Haw, An Oft-studied reaction that may never have been: Direct catalytic conversion of methanol or dimethyl ether to hydrocarbons on the solid acids HZSM-5 or HSAPO-34, J. Am. Chem. Soc., 124 (2002) 3844-3845.
[4] A.D. Chowdhury, K. Houben, G.T. Whiting, M. Mokhtar, A.M. Asiri, S.A. Al-Thabaiti, S.N. Basahel, M. Baldus, B.M. Weckhuysen, Initial Carbon–Carbon Bond Formation during the Early Stages of the Methanol-to-Olefin Process Proven by Zeolite-Trapped Acetate and Methyl Acetate, Angew. Chem. Int. Ed., 55 (2016) 5840-15845.
[5] R. Khare, S.S. Arora, A. Bhan, Implications of Cofeeding Acetaldehyde on Ethene Selectivity in Methanol-to-Hydrocarbons Conversion on MFI and Its Mechanistic Interpretation, ACS Catal., 6 (2016) 2314-2331.
Contact
Veronique Van Speybroeck
Kristof De Wispelaere