Machine learning of atomic polarizabilities for polarizable force field development
Machine learning of atomic polarizabilities for polarizable force field development
Promotor(en): T. Verstraelen /20MODEV04 / Model and software developmentProbleemstelling:
A fundamental understanding of experimentally observed material properties has in the last few decades originated more and more from atomistic computer simulations. These simulations are usually based on force fields to describe the interatomic interactions. In this regard, much interest has gone to the inclusion of electronic polarization effects, which stem from the deformation of the electronic wave functions due to the electric field generated by the surrounding molecules. The inclusion of polarization has proven to be vital in a wide range of intermolecular interactions. Models to include these effects often rely on the determination of atomic polarizabilities. These quantities are not uniquely defined, and thus require approximate models, which are often based on expensive quantum mechanical calculations. A promising differing approach is the use of machine learning. Such models can be trained to reproduce the ab-initio molecular polarizability from the geometry of the molecule or periodic structure of interest. The local nature of the network subsequently allows for a partitioning of the molecular polarizability into atomic fragments.
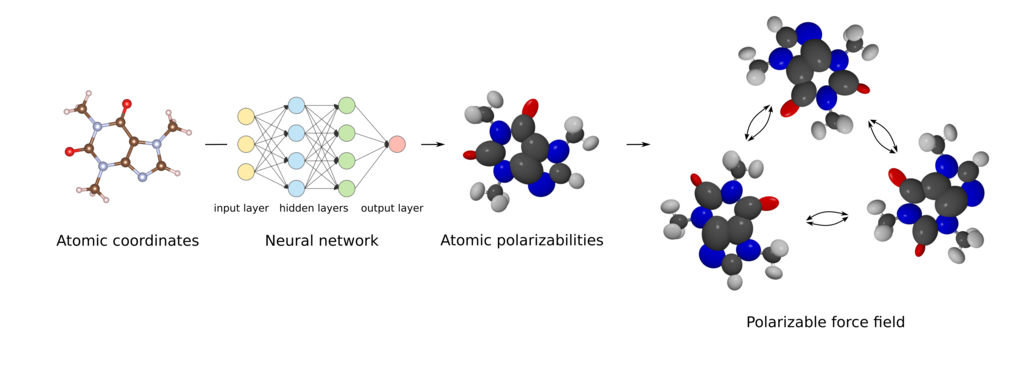
Figure 1. Machine learning approach to predicting atomic polarizabilities of atoms in molecules.
Doelstelling:
The first step in this thesis is constructing an appropriate machine learning model. Several ideas from the literature such as smooth overlap of atomic positions (SOAP) or SchNet can be used for this task.[1] A dataset of molecular polarizabilities (QMb7) is available in the literature, and will be used to train the model. However, a pure data-driven approach tends to result in atomic polarizabilities which are not appropriate for use in polarization models.[2] To overcome this, two differing approaches will be examined. In the first, a physical prior is included in the cost function of the model to ensure the prediction of accurate and useful atomic polarizabilities. In a second, the physical prior atomic polarizabilities are learned by the model first, after which they are modified to reproduce the molecular polarizability by means of delta learning. Several ideas for this prior physical insight, based on an ab-initio calculation of the molecule, have been proposed in the literature[3] and will be borrowed.
The resulting predicted atomic polarizabilities will be used in polarization models to benchmark the interaction energies between the strongly hydrogen-bonded molecules in the HBC6 dataset. This will serve as a demonstration of the quality of the predicted polarizabilities. Moreover, atomic polarizabilities are required in many dispersion models, which are a dominant interaction stabilizing many nonpolar complexes. Therefore, to test the performance of the predicted polarizabilities in these models, interaction energies of complexes in the S12L set of molecules dimers will be calculated. In this way, the superior performance of machine learned polarizabilities over more traditional methods can be shown.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]Keywordsmachine learning, Computational physics, polarizability, Force fieldsReferences
[1] K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko & K.-R. Müller (2017) SchNet: SchNet – A deep learning architecture for molecules and materials. The Journal of Chemical Physics, 148(24). https://doi.org/10.1063/1.5019779
[2] D. M. Wilkins, A. Grisafi, Y. Yang, K. U. Lao, R. A. DiStasio, M. Ceriotti (2016) Accurate molecular polarizabilities with coupled cluster theory and machine learning. Proceedings of the National Academy of Sciences of the United States of America, 116(9), 3401-3406. https://doi.org/10.1073/pnas.1816132116
[3] A. Tkatchenko, M. Scheffler (2009) Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Physical Review Letters, 102, 073005. https://doi.org/10.1103/PhysRevLett.102.073005
Contact
Toon Verstraelen