Influence of topology and operating conditions on the formation and growth of aromatics in zeolites
Influence of topology and operating conditions on the formation and growth of aromatics in zeolites
Promotor(en): V. Van Speybroeck, K. De Wispelaere /18NANO02 / Nanoporous materialsAromatics are ubiquitous in hydrocarbon chemistry in zeolites. During alcohol conversion into hydrocarbons they can play an active or deactivating role in the hydrocarbon pool mechanism and during the catalytic cracking of alkanes and alkenes they deactivate the catalyst. Detailed insights into the mechanisms driving the formation of deactivating molecules can give valuable design guidelines for the next generation of catalysts. Such materials should allow an optimal control of the product selectivity and catalyst lifetime.
Studies on the aromatic formation in zeolitic materials are rather limited and suggest several possible mechanisms, depicted in Figure 1. [1–3] Possible mechanisms include alkene oligomerization and cyclization and cycloaddition (Diels-Alder type) reactions. The dienes in such reactions might be the result of Prins type reactions, which recently have been reported for methanol-to-hydrocarbons (MTH) conversion.[4] Preliminary theoretical insights into the pathways for aromatics formations at Brønsted acid sites are obtained in the zeolite catalyst ZSM-5. It remains, however, unclear how aromatics formation occurs on Lewis acid sites (for example Ca oxide clusters or extraframework aluminium) and defects like silanol groups.
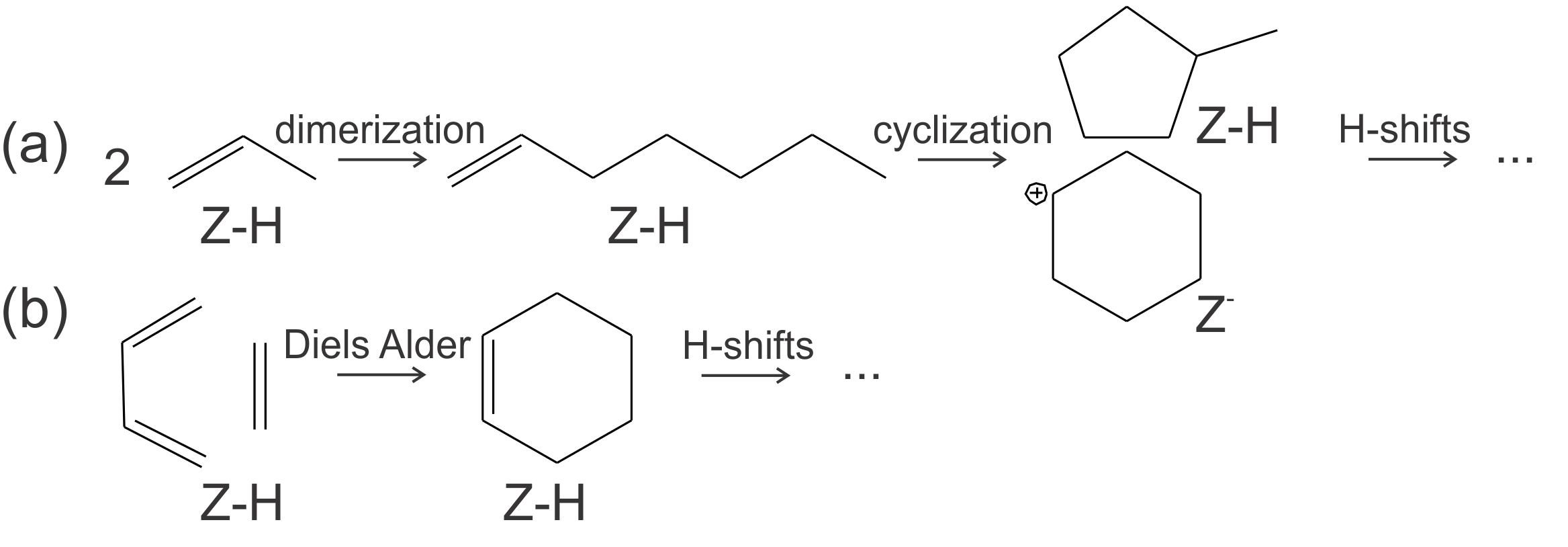
Figure 1 Schematic representation of the possible reaction mechanisms for the formation of aromatics, namely oligomerization and cyclization (a) and Diels-Alder type reactions (b).Moreover, detailed molecular-level insights into how operating conditions (temperature, pressure, feed composition) affect aromatics formation are lacking. Therefore, the relative importance of each proposed pathway should be investigated at different reaction conditions by constructing a DFT-based microkinetic model. Besides reaction conditions, steric effects induced by the zeolite topology can influence the stability and reactivity of the intermediates in the aromatics formation and thus the preferred pathway. Therefore, the study will be further extended by studying the aromatics formation in several 1D and 3D zeolite topologies, depicted in Figure 2, to acquire insight in how the shape and size of the pores might influence aromatics formation. It is well known that some zeolites can efficiently suppress the formation of aromatics[5] and more systematic insights into this effect are key to a further optimization of the catalyst.
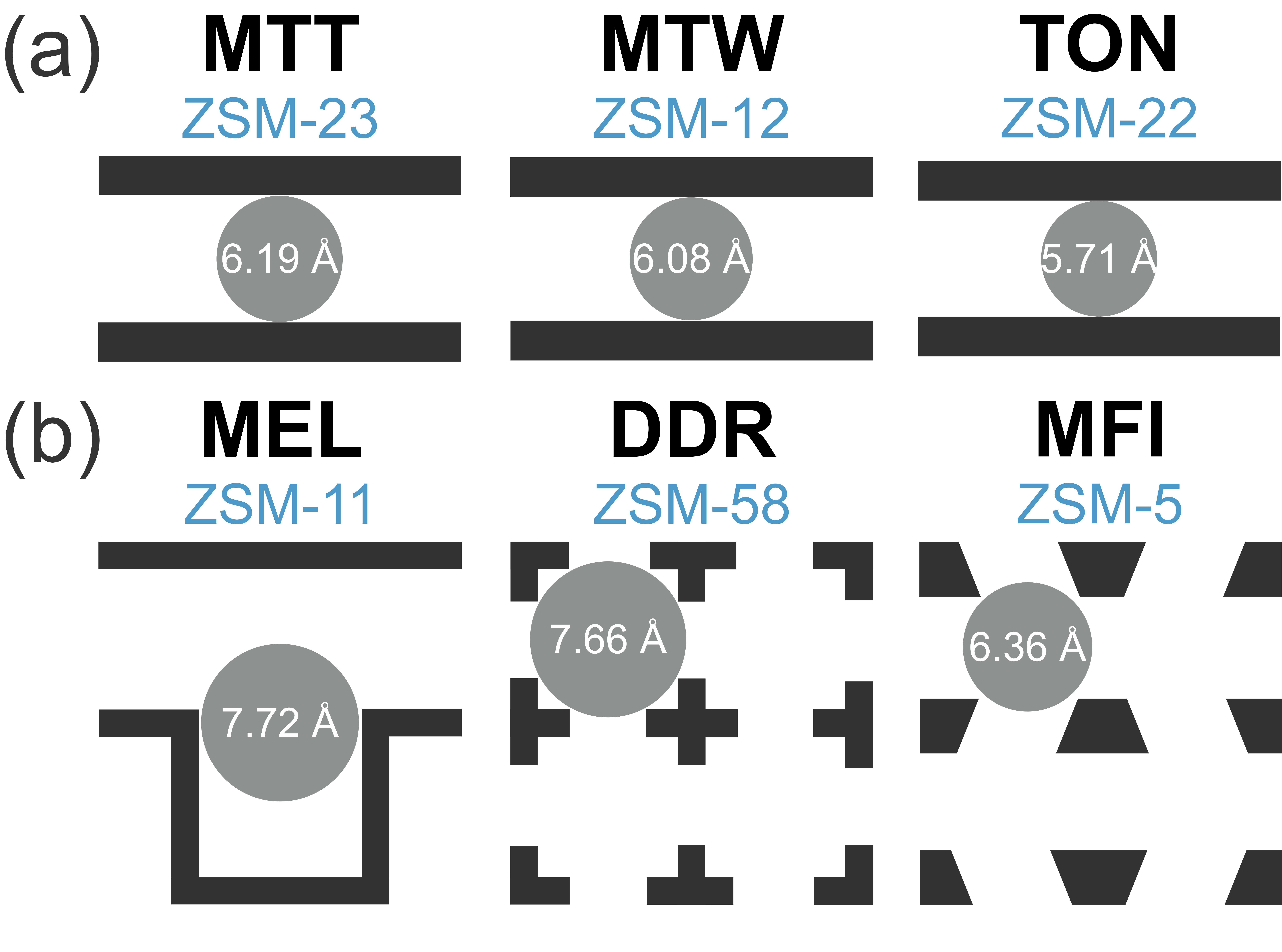
Figure 2 Schematic representation of the 1D (a) and 3D (b) zeolite topologies considered in this study.Goal
The objective of this master thesis is to study the influence of reaction conditions and catalyst topology on the relative importance of the proposed pathways towards aromatics formation at various types of active sites. To this end first principle chemical kinetics simulations will be performed on various elementary reactions leading to aromatic species. The calculated kinetics will then serve as input for a microkinetic model, which allows assessing the influence of operating conditions (temperature, pressure, feed composition) on the aromatics formation. By applying this methodology on catalyst models of ZSM-23, ZSM-12, ZSM-22, ZSM-11, ZSM-58 and ZSM-5, the influence of the zeolite topology will also be assessed. The molecular level insight obtained in this manner is crucial for an optimal catalyst design for alcohol conversion and catalytic cracking processes.
This project requires the application of several advanced molecular simulation techniques, which rely on first principle static and molecular dynamics simulations. The Center for Molecular Modeling has built up vast expertise in these advanced simulation techniques and collaborates on the subject with leading experimental and theoretical partners. The student will be actively coached to get acquainted with the plethora of techniques needed to tackle the proposed problem. It is the intention to involve the student actively in the work discussions with our collaborators. The CMM has access to sufficient computational resources to execute this research project. The proposed topic is challenging and requires technical skills, creativity and chemical insight.


- Study programmeMaster of Science in Chemical Engineering [EMCHEM]KeywordsHeterogeneous Catalysis, Chemical kinetics, Computational applicationsRecommended coursesMoleculaire modellering van industriële processen, Simulations and Modeling for the NanoscaleReferences
[1] M. Vandichel, D. Lesthaeghe, J. V. der Mynsbrugge, M. Waroquier, V. Van Speybroeck, J. Catal. 2010, 271, 67–78.
[2] K. D. Hungenberg, J. H. Kagon, B. E. Langner, J. Catal. 1981, 68, 200–202.
[3] N. Nikbin, S. Feng, S. Caratzoulas, D. G. Vlachos, J. Phys. Chem. C 2014, 118, 24415–24424.
[4] J.S. Martínez-Espín et al., J. Catal., 2017, 349, 136-148.
[5] S. Teketel, U. Olsbye, K.-P. Lillerud, P. Beato, S. Svelle, Microporous Mesoporous Mater. 2010, 136, 33–41.
Contact
Veronique Van Speybroeck
Kristof De Wispelaere