Including nuclear quantum effects via path integral techniques to accurately model breathing in metal-organic frameworks
Including nuclear quantum effects via path integral techniques to accurately model breathing in metal-organic frameworks
Promotor(en): V. Van Speybroeck /17NANO08 / Nanoporous materialsMetal-organic frameworks (MOFs) are a unique class of porous crystalline materials, which can easily be tuned both chemically and physically towards specific applications. These crystals consist of inorganic moieties connected by organic linkers. While nuclear quantum effects (NQEs) are of paramount importance to describe molecular interactions, as in most solid-state physics, they are almost never included in molecular simulations. However, a quantum treatment of the nuclear degrees of freedom, especially for the light atoms, is required to capture subtle effects such as quantum tunneling, deviations from the classical heat capacity due to energy quantization and zero-point effects on mechanical properties. An open question regarding this nuclear quantum effects is to which extent they alter the aforementioned properties.
The interest in modeling NQEs in atomistic simulations has been growing constantly over the past years. Including NQEs in simulations has become possible due to faster computers, but also due to the implementation of efficient algorithms in software packages such as i-PI [1]. One widely accepted technique is path integral molecular dynamics (PIMD), in which the Born-Oppenheimer approximation is used to separate the nuclear and electronic part of the wave function. In contrast with standard molecular dynamics (MD), where the positions and velocities of the atoms evolve in time by integrating the Newtonian equations of motion, the nuclei are treated quantum mechanically. This is done by mapping each nucleus onto a classical system of several fictitious particles – so-called beads – which are connected by harmonic springs. This is the classical isomorphism and the Hamiltonian governing these equations is derived from Feynman’s interpretation of quantum mechanics.

While exact in the limit of infinite beads, only a limited number of beads can be used in a real simulation. Several approaches have been developed to reduce the computational overhead by accelerating the convergence of properties with the number of beads. For example, 128 beads are required to obtain a good estimate of the heat capacity of water at room temperature, making these calculations 128 times more expensive than standard MD. In this project, we aim to use the “high-order path integral techniques”, a more accurate and computationally more efficient formulation of standard PIMD [2].
Recently, the Center of Molecular Modeling has built up a vast expertise in modeling flexible MOFs. An example of such a framework is MIL-53(Al), which displays ‘breathing’ behavior: the structure is able to expand (large-pore phase) or collapse (narrow-pore phase) under various thermodynamic stimuli such as temperature or pressure, in which the volume can change drastically with more than 35 procent. We have developed several methodologies to accurately describe this phenomenon with standard MD simulations [3].
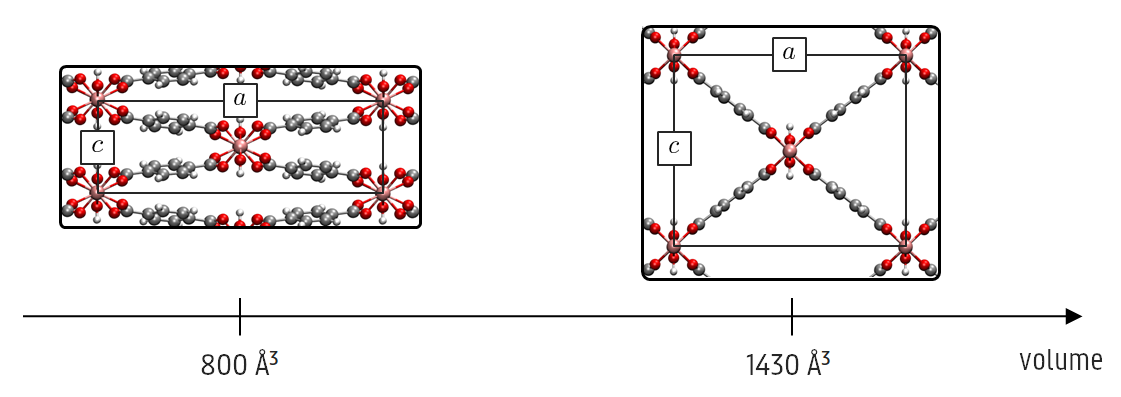
This work is highly relevant as the combination of PIMD and our existing methodology for modeling flexible materials has never been applied on MOFs or other nanoporous materials in general. The PIMD simulations will be carried out with force fields, in which the complex potential energy surface is approximated by simplified analytical expressions inspired by simple spring models. During the past few years, the CMM has built up a vast expertise in developing these force fields, resulting in a user-friendly force-field generator code, QuickFF [4]. The PIMD simulations itself will be carried out with i-PI.
Goals
In this thesis, the student will perform force-fields based PIMD to assess the importance of NQEs on the heat capacity and the mechanical behavior of flexible MOFs for the first time. The student will therefore have to study the relevant literature on path integral molecular dynamics, and implement and validate improvements on the existing methodology for modeling flexible MOFs. As such, this thesis bridges the gap between the physical fundamentals (including nuclear quantum effects via PIMD) and the model development to arrive at the engineering applications of MOFs.
The Center for Molecular Modeling has ample expertise in modeling nanoporous materials, and especially MOFs, and performs this research in the framework of large-scale international research programs. For these materials, various force fields were developed and applied to successfully predict a multitude of physical and chemical properties. The modeling is performed in close collaboration with both theoretical and experimental partners – including the research team of Ceriotti, the developer of i-PI – to achieve an optimal synergy between modeling and experimental efforts. The student will be actively involved in the work discussions with these international prominent partners. Furthermore, the student will be actively coached to get acquainted as soon as possible with all necessary techniques to perform the suggested research successfully.Context Engineering
Physics: investigate the influence of nuclear quantum effects on the mechanical behavior of flexible metal-organic frameworks.
Engineering: tune the available methodology for an optimal performance.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELLING, NANOKeywordsNuclear quantum effects, Thermodynamics, mechanical behavior, Molecular simulation, Nanoporous materialsReferences
1. M. Ceriotti, J. More and D.E. Manolopoulos, i-PI: A Python interface for ab initio path integral molecular dynamics simulations, Comp. Phys. Comm. 185, p. 1019-1026, 2014
2, V. Kapil, J. Behler and M. Ceriotti, High Order Path Integrals Made Easy, J. Chem. Phys. 145, 234103, 2016
3. S.M.J. Rogge, L. Vanduyfhuys, A. Ghysels, M. Waroquier, T. Verstraelen, G. Maurin and V. Van Speybroeck, A Comparison of Barostats for the Mechanical Characterization of Metal-Organic Frameworks, J. Chem. Theory Comput. 11, p. 5583-5597, 2015
4. L. Vanduyfhuys, S. Vandenbrande, T. Verstraelen, R. Schmid, M. Waroquier and V. Van Speybroeck, QuickFF: A program for a quick and easy derivation of force fields for Metal-Organic Frameworks from ab initio input, J. Comp. Chem. 36, p. 1015-1027, 2015
Contact
Veronique Van Speybroeck