High-throughput screening of promising covalent organic frameworks to design next-generation fuel cells
High-throughput screening of promising covalent organic frameworks to design next-generation fuel cells
Promotor(en): V. Van Speybroeck, S.M.J. Rogge /20NANO09 / Nanoporous materialsProbleemstelling:
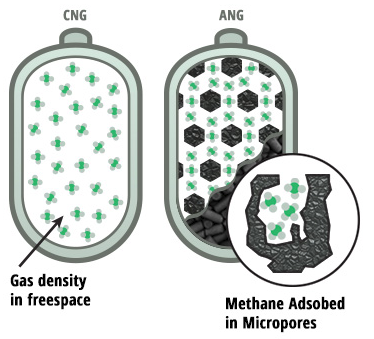
The recent discovery of a new class of nanoporous crystalline materials ushered in an intriguing new era in science. These materials – so-called covalent organic frameworks (COFs) – are easily tunable, allowing for a rich variety in chemical and physical properties. As COFs only consist of light-weight atoms that are held together by strong covalent bonds, they tend to combine a very low mass density with a high stability; an attractive combination that makes them of particular interest for a multitude of industrial applications. The storage of natural gas in COFs is such an application field that has attracted substantial research interest as natural gas forms a promising alternative to conventional petroleum-based fuels for transportation because of its abundant reserves and lower carbon emissions. To efficiently store natural gas, which mainly consists of methane, fuel cells are typically filled with nanoporous materials such as COFs, as illustrated in Figure 1. Owing to the favorable interactions between the gas and the material’s large internal surface, which can be tuned to a large extent, the COF-filled fuel tank can adsorb more natural gas in its pores than the empty vessel, resulting in a more efficient fuel cell [1].
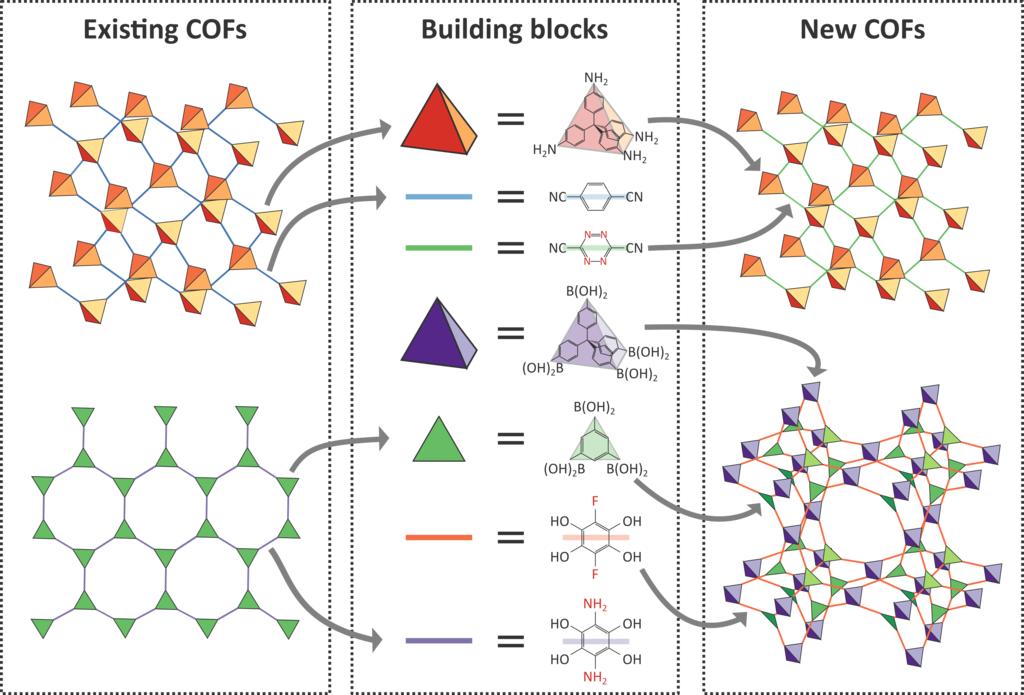
As the different building blocks in COFs can easily be extracted and recombined to generate yet-undiscovered COFs, an almost unlimited amount of hypothetical COFs could in principle be synthesized, as indicated in Figure 2. However, as the experimental characterization of these materials is time-consuming, it is not possible to investigate this large versatility of COFs experimentally. Instead, a computational screening is much more efficient and allows for the characterization of this vast search space of materials in a more feasible timeframe. This computationally-aided design therefore has the potential to hugely accelerate the discovery of promising COF structures with an extraordinary natural gas uptake for application in fuel cells for passenger vehicles [2]. In order to create COFs that can be implemented in industry, it is important to design materials that are sufficiently robust, so to withstand the thermal and mechanical stimuli to which the fuel tank is exposed during its lifecycle. By accurately optimizing critical parameters such as the thermal expansion, thermal conductivity, bulk modulus, and shear modulus during COF design, a sufficient thermal and mechanical stability will be ensured. Despite the fact that these parameters can be predicted through computer simulations, systematic knowledge on these physical properties for the relevant COFs and the influence of guests on these properties is still lacking [3].
Doelstelling:
The development of recent models at the Center for Molecular Modeling (CMM) has enabled an accurate description of adsorption in porous materials [4, 5] and allows for the efficient characterization of their thermal and mechanical stability [6, 7]. Until now, however, we mainly focused on the development of accurate techniques to study the intrinsic stability of the guest-free materials. This thesis aims to accurately characterize the mechanical and thermal properties of a rich variety of guest-loaded COFs. Furthermore, it is the intention to apply this computational protocol on those COFs that form promising candidates to be used in natural gas fuel tanks, thereby identifying those materials that are of particular interest to be applied in industrial setups.
To obtain the relevant adsorption, thermal and mechanical properties, various simulation techniques that will be taught during the thesis will be combined. These techniques will predominantly be based on Monte Carlo and molecular dynamics simulations or hybrid forms, considered in different ensembles [5]. To obtain accurate predictions of these properties, we will adopt in-house developed force fields fitted to quantum mechanical reference data [8]. The influence of the description of the interaction between the framework and the guest molecules on the various properties will have to be carefully evaluated.
The goal of the thesis is twofold. At first, we will benchmark our predictions on the well-known COF-5 and COF-102 for which abundant literature data is available. We will investigate the uptake capacity and the influence of adsorbed methane, ethane, nitrogen gas and carbon dioxide – the main components of natural gas – on the thermal and mechanical properties of the framework. In a second step, we will tap the large database of COF structures and corresponding force fields that were recently developed at the CMM based on the procedure highlighted in Figure 2. This large database will be investigated in a high-throughput fashion to identify those COF structures harboring a vast potential to take up large amounts of natural gas. The proposed high-throughput screening will already filter out the vast majority of hypothetical COFs that are unsuitable for fuel cells. Afterwards, the thermal and mechanical stability of the remaining structures will be investigated to ensure that they are sufficiently stable for industrial applications. Based on these insights, it is the aim to propose new pathways to tune the physical properties of COFs for fuel cell applications via the insights obtained in this thesis.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsCovalent organic frameworks, fuel cells, mechanical properties, Thermal properties, gas adsorption, high-throughputReferences
[1] H. Furukawa and O. M. Yaghi, “Storage of Hydrogen, Methane, and Carbon Dioxide in Highly Porous Covalent Organic Frameworks for Clean Energy Applications,” J. Am. Chem. Soc., vol. 131, no. 25, pp. 8875-8883, 2009.
[2] C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, “Large-scale screening of hypothetical metal–organic frameworks,” Nat. Chem., vol. 4, pp. 83-89, 2012.
[3] S. R. G. Balestra, R. Bueno-Perez, S. Hamad, D. Dubbeldam, A. R. Ruiz-Salvador en S. Calero, „Controlling Thermal Expansion: A Metal–Organic Frameworks Route,” Chem. Mater., vol. 28, nr. 22, pp. 8296-8304, 2016.
[4] S. Vandenbrande, T. Verstraelen, J. J. Gutiérrez-Sevillano, M. Waroquier and V. Van Speybroeck, “Methane Adsorption in Zr-Based MOFs: Comparison and Critical Evaluation of Force Fields,” J. Phys. Chem. C, vol. 121, no. 45, pp. 25309-25322, 2017.
[5] S. M. J. Rogge, R. Goeminne, R. Demuynck, J. J. Gutiérrez-Sevillano, S. Vandenbrande, L. Vanduyfhuys, M. Waroquier, T. Verstraelen en V. Van Speybroeck, „Modeling Gas Adsorption in Flexible Metal–Organic Frameworks via Hybrid Monte Carlo/Molecular Dynamics Schemes,” Adv. Theory Simul., vol. 2, p. 1800177, 2019.
[6] L. Vanduyfhuys, S. M. J. Rogge, J. Wieme, S. Vandenbrande, G. Maurin, M. Waroquier and V. Van Speybroeck, “Thermodynamic insight into stimuli-responsive behaviour of soft porous crystals,” Nat. Commun., vol. 9, p. 204, 2018.
[7] S. M. J. Rogge, M. Waroquier en V. Van Speybroeck, „Reliably Modeling the Mechanical Stability of Rigid and Flexible Metal–Organic Frameworks,” Acc. Chem. Res., vol. 51, nr. 1, pp. 138-148, 2018.
[8] L. Vanduyfhuys, S. Vandenbrande, J. Wieme, M. Waroquier, T. Verstraelen and V. Van Speybroeck, “Extension of the QuickFF force field protocol for an improved accuracy of structural, vibrational, mechanical and thermal properties of metal–organic frameworks,” J. Comput. Chem., vol. 39, no. 16, pp. 999-1011, 2018
Contact
Veronique Van Speybroeck