High-throughput screening to predict the textural and adsorption properties of covalent organic frameworks using a top-down approach
High-throughput screening to predict the textural and adsorption properties of covalent organic frameworks using a top-down approach
Promotor(en): V. Van Speybroeck /17MODEV10 / Model and software developmentCrystalline, nanoporous materials are inherently present in several day-to-day applications such as energy storage, gas separation, and the capture of greenhouse gases. Their unique combination of long-range order and tunable pores makes these materials tractable for a variety of applications relying on high surface areas, high porosities, and chemical versatility, such as guest adsorption. Thanks to favorable interactions between these materials and the adsorbed gas, the amount of gas that can be stored inside the material is larger than in an empty recipient with the same volume. Covalent organic frameworks (COFs) form a recently proposed class of these materials, consisting solely of organic moieties connected by strong covalent bonds [1]. A COF’s structure can be decomposed into secondary building units (SBUs): simplified polyhedra consisting of tens of atoms (see top pane of Figure 1). SBUs extracted from different COFs can then be combined – not unlike building K’NEX structures – forming a new, hypothetical material, a process which is called reticular chemistry (see bottom pane of Figure 1) [2].
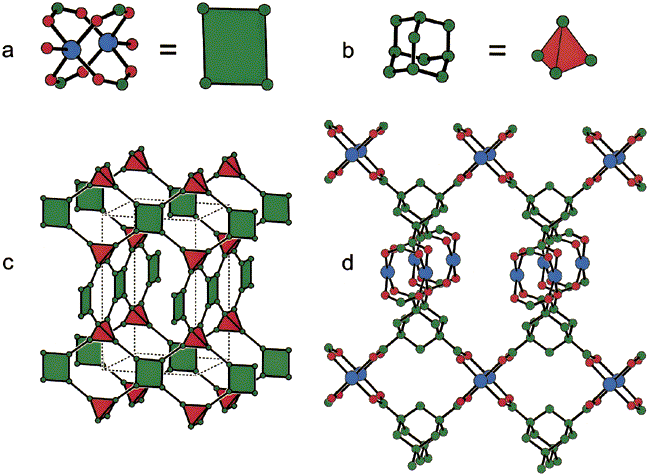
Thanks to the large amount of potential SBUs, the variety of possible topologies to build the scaffold, and the ability to introduce extra active functionalities to the material by post-functionalization, a large versatility of COFs arises. However, experimentally synthesizing all these COFs would be a daunting task. Ideally, the various degrees of freedom in constructing these materials may be controlled and fine-tuned at the molecular level, yielding materials with the proper structural, electronic, and functional characteristics for a given application. From this, phenomenological rules can be established to identify a smaller subset of these materials performing well for the given application. Model-guided design of these materials in a high-throughput way, made possible thanks to the principles of reticular chemistry, is hence an urgent topic to advance this field.
Goal
The aim of this thesis is twofold. In a first step, a procedure needs to be defined to build a computational model of all possible COFs given a certain topology and a certain set of SBUs: the so-called top-down approach in high-throughput screening [3]. This procedure can be inspired by similar work on metal-organic frameworks, such as the weaver Pythonic code developed by our collaborators at the Ruhr-University of Bochum [4]. In this procedure, the topology needs to be deconstructed into vertices (which will later correspond to a SBU) and edges (which corresponds to the SBU interconnectivity). For this, the possible points of extension of each SBU – i.e. the points where the SBU can be connected by SBUs – need to be defined [2]. Subsequently, for each SBU with the correct number and orientation of points of extension, a hypothetical COF will be constructed and optimized, for instance using a computationally efficient semi-empirical method. As output, this procedure will yield all hypothetical, stable COFs with a predefined topology and composed of the inputted subset of the SBUs.
In a second step, properties of the created hypothetical COFs need to be analyzed. Using existing programs, structural properties such as cell lengths and stacking order, and textural properties such as pore sizes and accessible void fraction can then be extracted. Moreover, using Grand Canonical Monte Carlo (GCMC) simulations, the adsorption of small molecules such as methane and carbon dioxide in the stable COFs will be studied, specifically determine the favorable adsorption sites and the amount of guests than can be adsorbed (see for instance Figure 2) [5]. By analyzing the correlations between the structural and textural properties on the one hand and the adsorption properties on the other hand, structure-property relationships can be established. Moreover, those parameters which are found to describe best the guest adsorption can be extracted, and used to represent the set of materials in a low-dimensional space, with these parameters as descriptors. Using data mining techniques borrowed from the data science community, such as support vector machines, these relationships can be analyzed systematically and the observed correlations can be transferred to other hypothetical COFs. Finally, the construction of decision trees will advance further research in this area, giving clear guidelines to experimentalists which COFs are expected to perform best for a given application, such as methane adsorption.
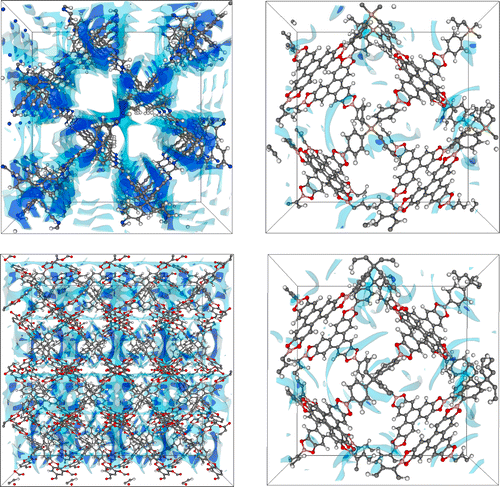
The student will be actively coached to make him/her acquainted with the advanced simulations techniques early in the thesis year, and to transfer necessary programming skills needed to perform the research. A strong interest in programming is, however, a prerequisite.
Aspects
Physics aspect: Adsorption of guest molecules; identifying the relevant parameters to maximize adsorption
Engineering aspect: High-throughput screening procedure to select best in class materials.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELLINGKeywordsImplementation, High-throughputberekeningen, data mining, data analysis, guest adsorption, Covalent organic frameworksReferences
[1] A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, “Porous, crystalline, covalent organic frameworks,” Science, vol. 310, no. 5751, pp. 1166-1170, 2005.
[2] M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, "Modular chemistry: Secondary building units as a basis for the design of highly porous and robust metal-organic carboxylate frameworks," Acc. Chem. Res., vol. 34, no. 4, pp. 319-330, 2001.
[3] Y. J. Colón and R. Q. Snurr, "High-throughput computational screening of metal-organic frameworks," Chem. Soc. Rev., vol. 43, no. 16, pp. 5735-5749, 2014.
[4] S. Amirjalayer, R. Q. Snurr and R. Schmid, "Prediction of structure and properties of boron-based covalent organic frameworks by a first-principles derived force field," J. Phys. Chem. C, vol. 116, no. 7, pp. 4921-4929, 2012.
[5] R. L. Martin, C. M. Simon, B. Medasani, D. K. Britt, B. Smit and M. Haranczyk, "In silico design of three-dimensional covalent organic frameworks via known synthesis routes and commercially available species," J. Phys. Chem. C, vol. 118, no. 41, pp. 23790-23802, 2014.
Contact
Veronique Van Speybroeck