First-principle kinetic study on the methylation of hydrocarbons in small-, medium- and large-pore zeolitic materials
First-principle kinetic study on the methylation of hydrocarbons in small-, medium- and large-pore zeolitic materials
Promotor(en): V. Van Speybroeck /15_NANO09 / Nanoporous materialsDepleting oil reserves ignited the interest in the conversion of methanol to hydrocarbons (MTH) or olefins (MTO). This conversion takes place in zeolite and zeotype materials as their nanoporous channels and cages impose selectivity to the process. It has been found that the inorganic catalyst framework and occluded hydrocarbons catalyzed the MTO reactions. Moreover, a couple of catalytic cycles operate simultaneously during olefin formation, resulting in a very complex reaction mechanism which is currently still not fully understood. Herein, molecular simulations are an indispensable tool as they can provide molecular-level insights.
Within this complex mechanism, methylation reactions of hydrocarbons are crucial steps, since it has been proven that they initiate every active cycle of the MTO process. In the early stages of the process, an equilibrium mixture of methanol, dimethyl ether (DME) and water is formed. Hence, methylations reactions can start from methanol or DME. Moreover, two methylation mechanisms have been proposed. In the concerted reaction step, the methylated product and a water molecule are formed simultaneously whereas the reaction goes through a methoxide intermediate in the stepwise mechanism (Figure 1). Depending on the catalyst topology, the material’s acid strength and the process conditions the relative importance of both mechanisms differs. The latest experimental insights show that the prevailing mechanism at low and higher temperature is different due to variations in the entropic barrier. However to date a detailed understanding of the factors governing this key reaction step is not clear yet.
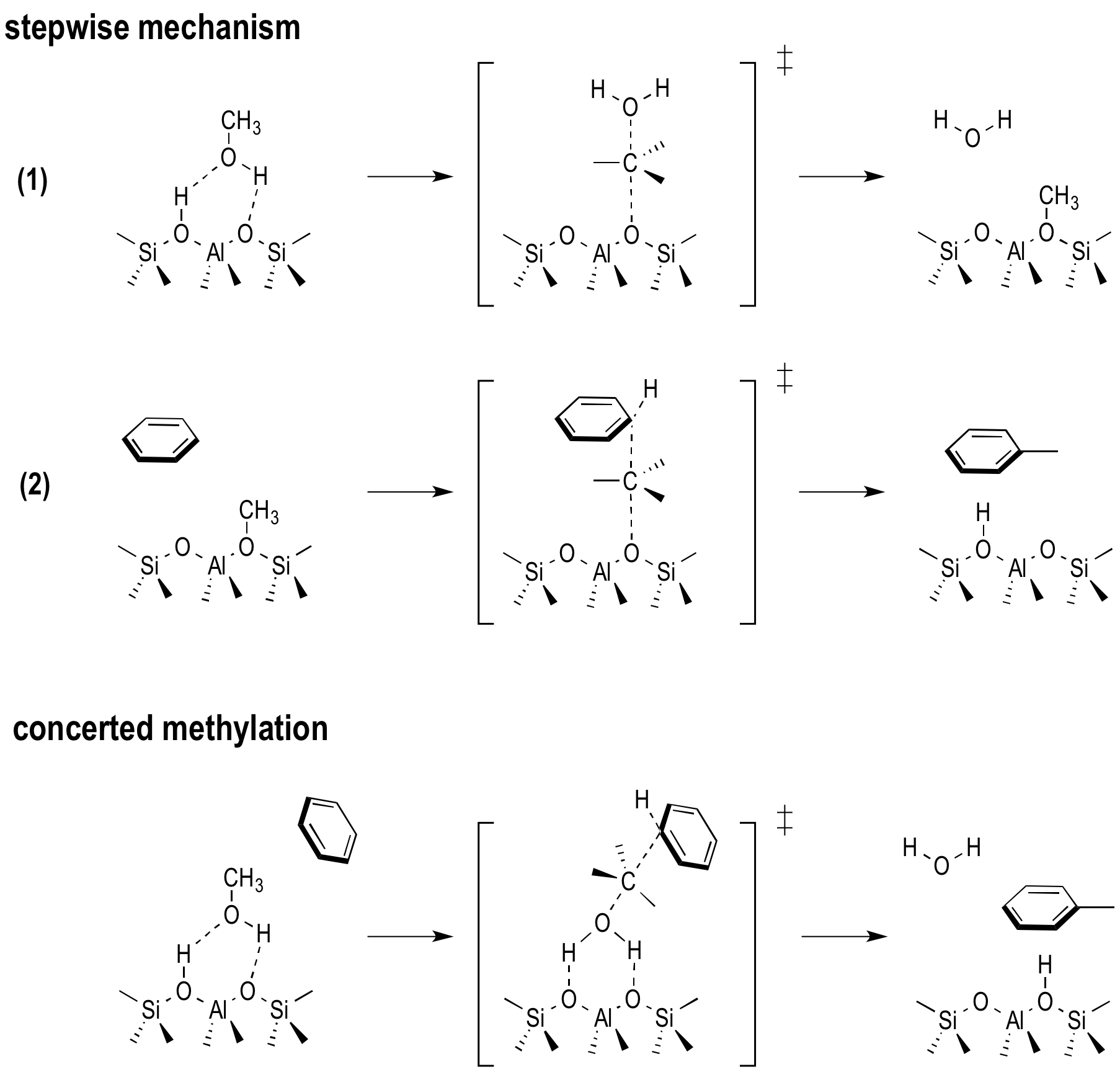
Figure 1. Stepwise and concerted reaction mechanism for the methylation of benzene.Goal In this master thesis we want to investigate the the competition between the stepwise and concerted methylation under the influence of the size of the zeolitic pores, the acid strength and the process conditions. Instead of using 0 K simulations, we will perform advanced molecular dynamics simulations where the the competition between two mechanisms can be sampled in one single run and we will be able to reconstruct the free energy at operating conditions (Figure 2). This will all be done at realistic temperatures, pressures, account for the framework flexibility and the presence of multiple guest molecules. The research will be performed in the framework of a larger concerted action to simulate chemical transformations at operating conditions. The Center for Molecular Modeling has built up vast expertise in these advanced simulation techniques and collaborates on the subject with leading experimental and theoretical partners. It is the intention to involve the student actively in the work discussions with our collaborators. The CMM has access to sufficient computational resources to execute this research project. The proposed topic is challenging and requires technical skills, creativity and chemical insight. Furthermore there will be a close interaction with the researchers working on spectroscopy in nanoporous materials (headed by Prof. K. Hemelsoet).
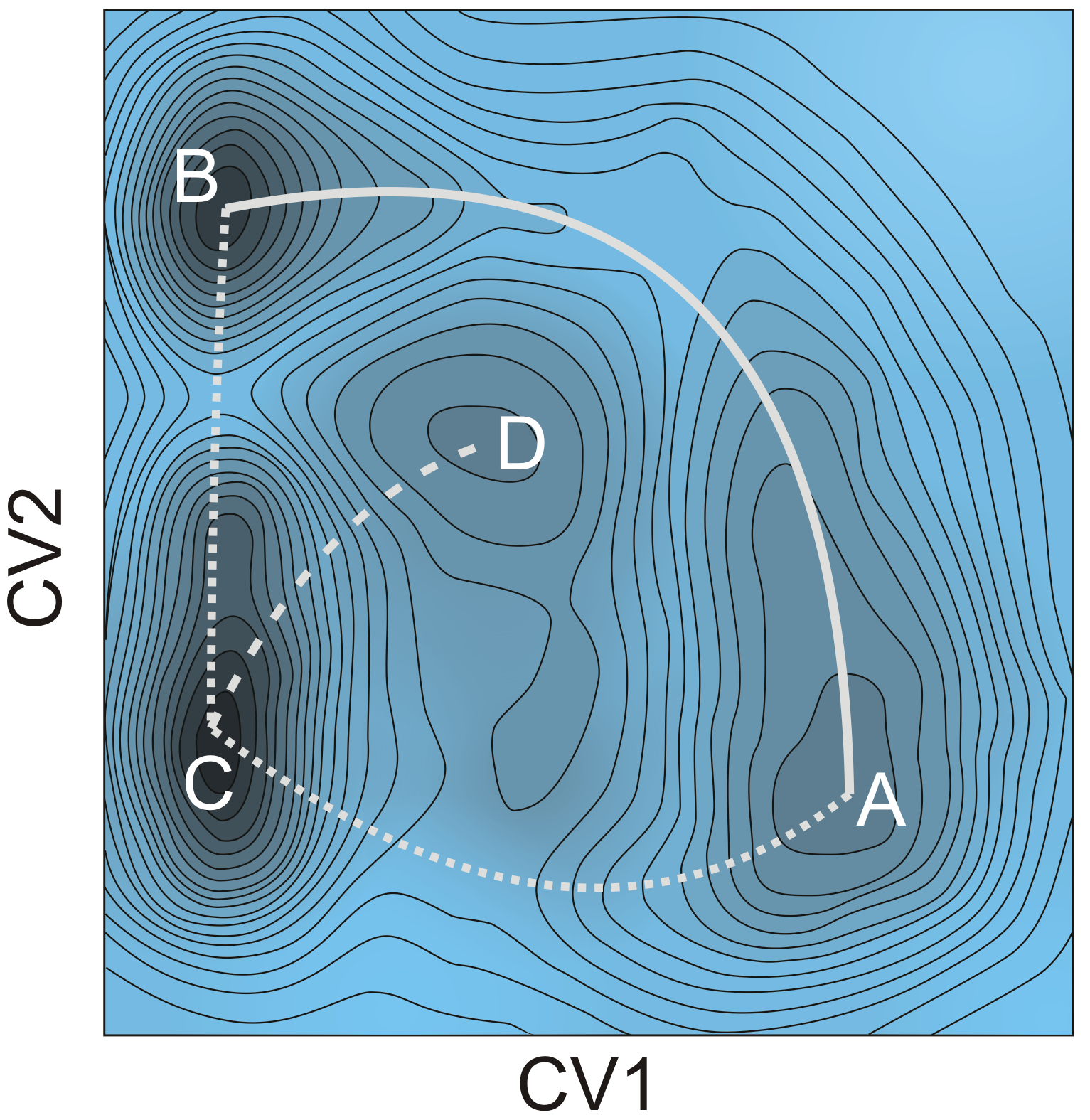
Figure 2 : Schematic representation of a complex 2D free energy surface with indication of several reaction pathways and intermediates.


- Study programmeMaster of Science in Chemical Engineering [EMCHEM], Master of Science in Chemistry [CMCHEM]KeywordsHeterogeneous Catalysis, Chemical kinetics, Computational applications
Contact
Veronique Van Speybroeck
Kristof De Wispelaere