Extracting kinetic information from advanced computer simulations to investigate the diffusion of protic molecules through small-pore zeolites
Extracting kinetic information from advanced computer simulations to investigate the diffusion of protic molecules through small-pore zeolites
Promotor(en): V. Van Speybroeck /18NANO13 / Nanoporous materialsNanoporous materials such as zeolites and metal-organic frameworks are attractive for a broad variety of industrial applications, ranging from chemical and physical sensing, over gas adsorption and separation, to heterogeneous catalysis. The structural porosity of these materials instills in them the potential to efficiently envelop specific adsorbed guest molecules, as indicated in Figure 1, whereas their periodic framework allows for the inclusion of functional groups that preferentially interact with certain molecules. It is therefore not surprising that zeolites, which are composed of corner-sharing SiO4 and AlO4 tetrahedra forming scaffold-like networks, are omnipresent in today’s industrial applications and have received considerable attention from both experimentalists and computational physicists. Most prominently, these zeolites have been commercial-ized as catalysts in the methanol-to-olefins (MTO) process. In this process, methanol, which is extracted from natural gas or coal, is converted into longer olefins such as ethylene and propylene inside the pores of the material [1]. Once extracted from the pores, these olefins form the basic products for a variety of petrochem-ical end products such as plastics and pharmaceutical drugs. The MTO process has received a lot of attention in the last decade, as it forms an important green alternative to traditional processes starting from crude oil.
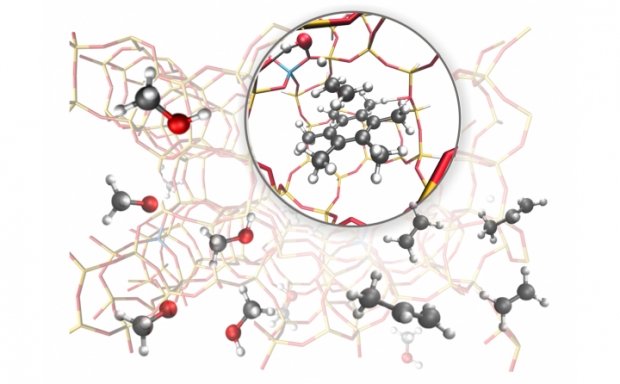
To optimize the efficiency of the MTO process in a zeolite, controlling diffusion inside the material plays a dominant role. First of all, the porous network of the zeolite should allow for the diffusion of methanol from the surface of the zeolite to the internal active sites where the conversion towards olefins take place. Second, given that multiple unwanted byproducts are formed in this process, the zeolite should allow for the selective diffusion of the olefins from the active sites to the surface, while limiting the diffusion of the unwanted by-products. For the MTO process, H-SAPO-34 has emerged as one of two industrially important zeolites. As indi-cated in Figure 2, this zeolite consists of large cages, in which the MTO conversion takes place, that are con-nected through substantially smaller windows. As these windows regulate the diffusion process, it is essential to understand from an atomistic point of view the diffusion of the small molecules of interest – methanol, ethylene, and propylene – through these windows. While computational modeling has already proven to be an indispensable tool to predict the species formed during the MTO process [1], it is still not fully understood which factors play a decisive role in the diffusion of methanol and other alcohols through the windows of the zeolite. However, microscopic insight in the diffusion of these small molecules is imperative for the further design of zeolites as catalysts in the industrial MTO process.
Objective
The objective of this master thesis is a systematic investigation of the diffusion of methanol and other alco-hols in H-SAPO-34 at operational conditions of temperature and pressure. If successful, the student will achieve insight into the molecular factors governing diffusion during the MTO process that will further guide the rational design of functional zeolites.
From a computational perspective, diffusion can be modelled by tracking the position of the molecule under study during the simulation. However, as diffusion is a phenomenon that inherently takes place at extended length and time scales, it is necessary to approximate the physical interactions between the atoms in the sys-tem by simplified analytical interactions via the introduction of classical force fields. These force fields should, however, still properly include the inherent flexibility of the framework at the elevated simulation tempera-tures in order to correctly model diffusion [2]. The Center for Molecular Modeling (CMM) has a long-standing collaboration with prof. German Sastre (Universitat Politècnica de València) that focusses on the development and application of these flexible force fields for zeolites. In this thesis, these existing zeolite force fields will be extended to account for the intermolecular interactions between methanol and the other guest molecules present in the zeolite. Therefore, a first objective of this thesis is the extension of current zeolite force fields with new potential energy terms for the adsorbed guest molecules, relying on existing force fields published in literature. Special attention should be paid to the correct modeling of the weak dispersion and electrostatic interactions between the different guest molecules and between the guest molecules and the framework, which play an important role during diffusion.
To model the diffusion of molecules through the small windows of H-SAPO-34, as visualized in Figure 3, a sec-ond challenge should be overcome. As molecules need to surmount appreciable energy barriers to diffuse through these small-pore windows, diffusion becomes a rare event. As a result, conventional molecular dy-namics (MD) simulations can no longer be employed. Therefore, enhanced sampling techniques such as metadynamics need to be adopted to describe the hopping process from one cage to the other. However, as these enhanced sampling techniques also distort time information by speeding up the occurrence of rare events, dedicated techniques are required to obtain the true diffusion coefficient [4]. In this thesis, we will investigate how concepts from statistical physics may be adopted to extract kinetic information from these enhanced simulations.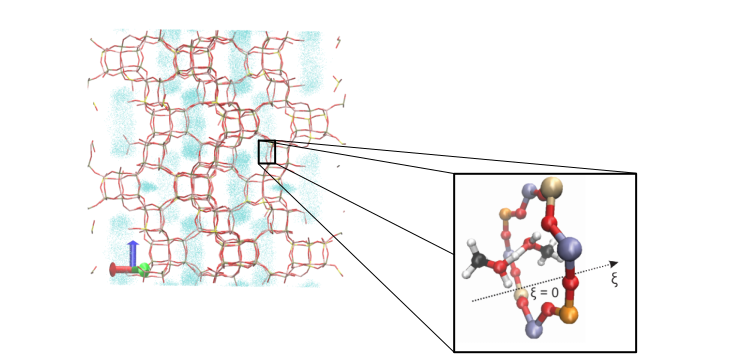
This master thesis, on the crossroads between non-equilibrium thermodynamics, statistical physics and com-putational physics, fits into a larger concerted action in which physical and chemical transformations in mi-croporous materials are computationally investigated. As the CMM collaborates with leading national and international partners in the frame of this program, such as prof. German Sastre, it is the intention to involve the interested student in our work discussions with our collaborators. The student will be made familiar with the simulation techniques necessary for this research proposal early on in the thesis, given the expertise avail-able at the CMM.
Aspects
Physics aspect: Use of classical mechanical models for materials modeling and the extraction of thermody-namic properties
Engineering aspect: Investigation of the MTO process for industrial purposes



- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELINGKeywordsMolecular dynamics, Force fields, Free energy, Nanoporous materialsReferences
[1] V. Van Speybroeck, K. De Wispelaere, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet and M. Waroquier, "First Principle Chemical Kinetics in Zeolites: The Methanol-to-Olefin Process as a Case Study," Chem. Soc. Rev., vol. 43, no. 21, pp. 7326-7357, 2014.
[2] R. V. Awati, P. I. Ravikovitch and D. S. Sholl, "Efficient and Accurate Methods for Characterizing Effects of Framework Flexibility on Molecular Diffusion in Zeolites: CH4 Diffusion in Eight Member Ring Zeolites," J. Phys. Chem. C, vol. 117, no. 26, pp. 13462-13473, 2013.
[3] P. Tiwary and M. Parrinello, "From Metadynamics to Dynamics," Phys. Rev. Lett., vol. 111, no. 23, p. 230602, 2013.
Contact
Veronique Van Speybroeck