Enthalpy or entropy? How temperature changes the dynamics of alkene cracking intermediates via ab initio derived machine learning potentials
Enthalpy or entropy? How temperature changes the dynamics of alkene cracking intermediates via ab initio derived machine learning potentials
Promotor(en): V. Van Speybroeck /28136 / Nanoporous materialsBackground and problem
Catalytic alkene cracking on zeolites plays an important role to shift and improve the product distribution toward a higher yield of light olefin or fuels in both classical petrochemical processes as well as emerging alternatives such as methanol-to-olefins (MTO), CO2 hydrogenation or biomass conversion. Mechanistically, alkene cracking proceeds through a complex reaction network of protonations, hydride transfers, isomerizations, oligomerizations, β-scissions, etc. [1,2] Within such a complex ensemble of elementary processes, multiple key intermediates like surface alkoxide species and carbenium ions can be formed [1]. Modelling these species is very challenging, as their stability is dictated by a delicate balance between enthalpic and entropic effects, the weight of the latter growing linearly with temperature. The stability of these intermediates is critically determined by the topology of the zeolite framework, the zeolite acidity and the reaction conditions. Nevertheless, proper insight into the stability and reactivity of the cracking intermediates is essential for improving the selectivity towards the desired products and for reducing the rapid aromatics formation and deactivation.
Previous modeling studies often focused either on very accurate static calculations of a single configuration, leading to precise estimates of the enthalpic contribution, while producing large errors on the entropy. On the other hand, some studies applied dynamic techniques which explicitly account for temperature effects and mobility of the intermediates, but due to their high cost, only approximate methodologies can be used and the accuracy of the enthalpic constribution is often questionable [3,4]. Presently, also machine learning potentials (MLPs) have entered the toolbox of computational chemists, opening the doors towards new and previously unaccessible opportunities. A properly trained MLP with ab initio data can be applied to compute molecular interactions and reaction energies with high accuracy at a cost reduced by several orders of magnitude. This allows to perform enhanced sampling simulations at any temperature of interest in order to explicitly assess the separate enthalpic and entropic contributions to the thermodynamic and kinetic stability of the reaction intermediates. The use of MLPs may hence provide better insight into the dynamic behavior and reactivity of the prevailing intermediates in alkene cracking as a function of the reaction conditions and zeolite properties.
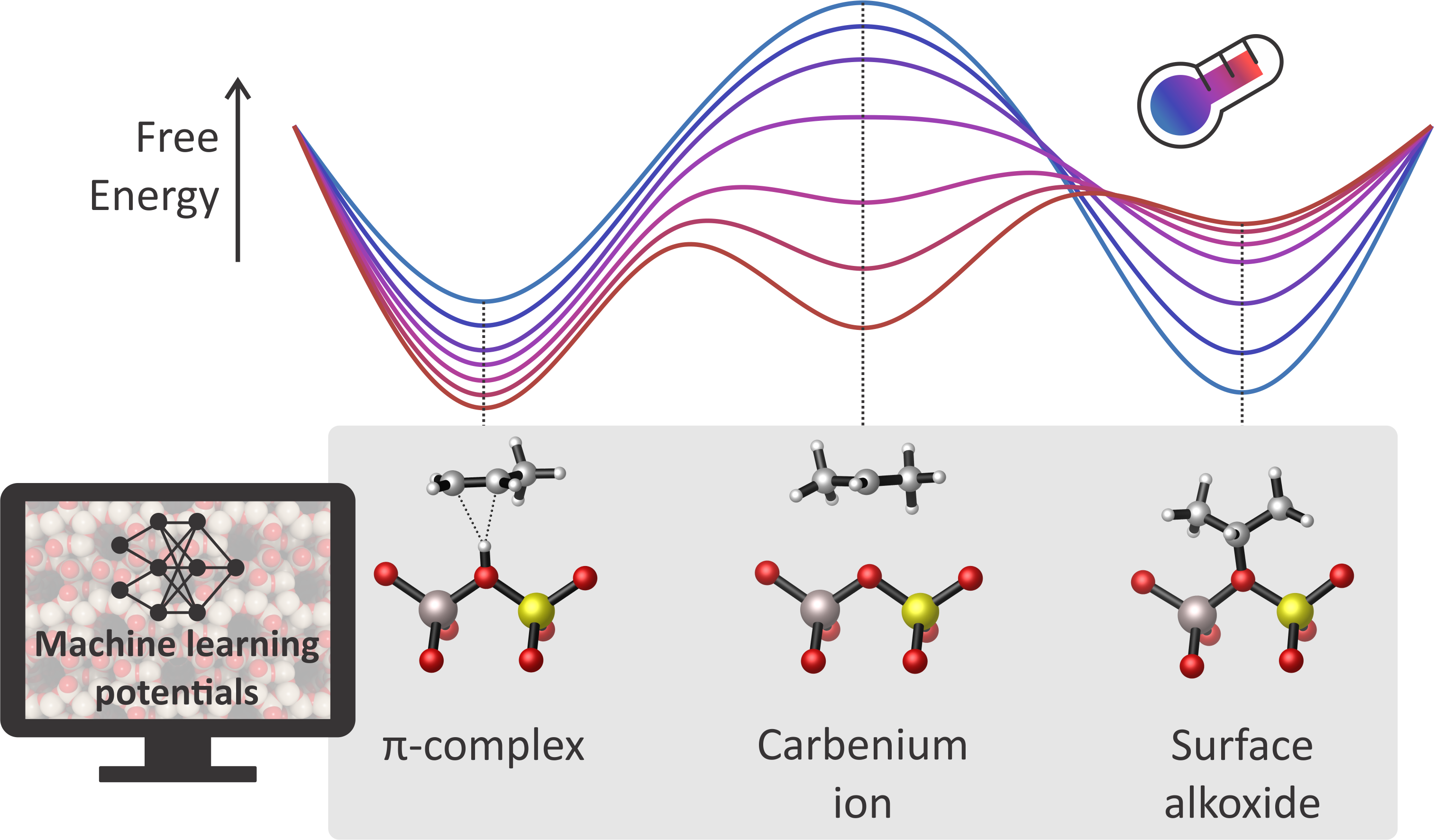
Figure 1: Machine learning potentials allow to unravel the stability of important alkene cracking intermediates at multiple temperature, providing fundamental insights for a theory-driven optimization of the process.
Goal
This master thesis’ objective is to unravel how reaction conditions (temperature, loading, zeolite acidity,…) affect the reactivity and stability of key intermediates in zeolite-catalysed alkene cracking, a still open matter of debate in current literature. To achieve this goal, you will start by performing ab initio simulations to investigate the adsorption and interconversion between different intermediates in various zeolite frameworks. Secondly, a subset of the generated structures will be used to train an accurate MLP, with which you will be able to extensively investigate the intermediates stability at multiple temperatures, explicitly deriving enthalpic and entropic contributions as a function of the framework topology. Finally, you will apply the obtained insights to study key elementary reaction steps with enhanced sampling simulations. Ultimately, the outcome of the simulations will result in an accurate assessment of the stability and reactivity of alkene cracking intermediates for various zeolite frameworks and reaction conditions which will assist in identifying the crucial parameters for increasing the catalyst selectivity and lifetime.
This project requires the application of several advanced molecular simulation techniques. The Center for Molecular Modeling has built up vast expertise in these techniques and collaborates on the subject with leading experimental partners. The student will be actively coached to get acquainted with the simulation techniques needed to tackle the proposed problem.
- Study programmeMaster of Science in Chemical Engineering [EMCHEM]KeywordsHeterogeneous Catalysis, Zeolites, Alkene cracking, Operando modelingReferences
[1] Cnudde, P. et al. ACS Catal. 2018, 8, 9579-9595.
[2] Standl, S. et al. Ind. Eng. Chem. Res. 2019, 58, 18107-18124.
[3] Ren, Q. et al. J. Phys. Chem. C 2020, 124, 10067-10078.
[4] Rey, J. et al. Angew. Chem. Int. Ed. 2020, 132, 19100-19104.
Contact
Veronique Van Speybroeck
Pieter Cnudde