Diffusion of mixtures of protic molecules and hydrocarbons through small pore zeolites
Diffusion of mixtures of protic molecules and hydrocarbons through small pore zeolites
Promotor(en): V. Van Speybroeck, A. Ghysels /17NANO11 / Nanoporous materialsNanoporous materials are structurally ordered materials that possess channels and pores of molecular dimensions, which makes them very tractable for various applications such as separation processes, gas storage, ... Zeolites are omnipresent in a wide variations of industrial applications. The materials are tecto-silicates, built out of SiO4 and AlO4 tetrahedra linked into corner-sharing networks. The function of these materials relies to a large extent on the diffusion of small molecules through the channel and pore system as schematically shown in Figure 1. Within the Center for Molecular Modeling we have ample expertise in studying the methanol to olefin conversion, a process where a methanol source is converted into olefins which may then be used to produce plastics,… This process received a lot of attention the last decade, as it is an important alternative to bypass crude oil. One of the crucial factors into the overall process is the diffusion of both incoming methanol molecules as produced small hydrocarbons such as propene which are produced during the MTO process. It is very difficult to understand the molecular factors governing these diffusion paths solely from experimental point of view. Molecular simulations are indispensable to describe the diffusion behaviour. So far we have studied the diffusion of ethene and propene in various types of zeolites. Both molecules are apolar and aprotic species, which interact with the walls of the zeolite via weak long range dispersion forces. The diffusion paths of methanol are expected to be more complicated, since they are able to make hydrogen bonds with the framework.
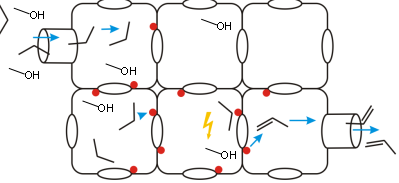
Goal
The objective of the master thesis is to study the diffusion of alcohols and more complex feedstocks through small pore zeolites via advanced computer simulations.
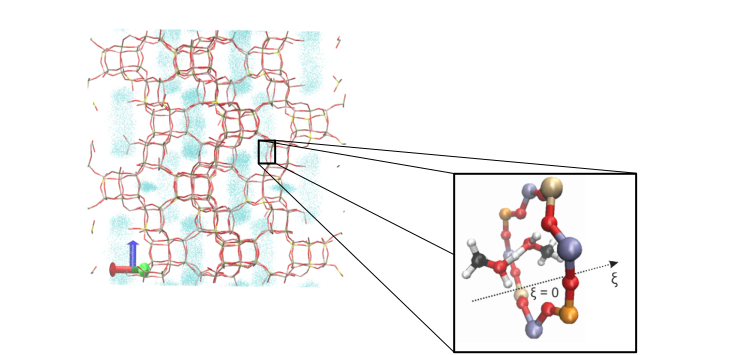
Diffusion is a phenomenon that takes place at larger length and time scales. It is typically studied using classical force field based approaches. In molecular dynamics simulations the position of the molecule is tracked over time, from which the diffusivity can be derived, as shown for propene in Figure 2. In some cases, molecules have to surmount energy barriers during their paths through the crystal. Diffusion then becomes a rare event, and special sampling techniques need to be used to describe the hopping process from one cage to the other.
For the diffusion of hydrocarbons in zeolites, the flexible behaviour of the zeolite rings is very influential [1]. In literature, several force fields were proposed to describe the flexibility of the zeolites (for example [2]). However for the intramolecular interaction of methanol and more complex feedstock, new interaction terms should be introduced. Thus a first objective is the extension of the force field based description with new potential energy terms able to describe the more complex feedstock intramolecular interactions. To this end we will rely on our force field based engine developed at the Center for Molecular Modeling, namely QuickFF. The new force field terms are derived from performing some ab initio quantum mechanical based simulations on smaller systems. Moreover, an important contribution to the force field describing the diffusion stems from the intermolecular interactions, describing the interaction between the zeolite and the guest molecules. Those non-bonding terms have to be described using appropriate electrostatic and Van Der Waals parameters.
Once the force field has been extended molecular simulations such as molecular dynamics and Monte Carlo simulations can be performed with our in-house developed force field code YAFF or using the DL_POLY code. As a result, simulations up to a few nanoseconds are possible, which are sufficient to accurately determine the mean-squared displacement (MSD) of the guest molecules, and hence its diffusion coefficient. Nevertheless, for large guest molecules the diffusion is severely hindered by the small windows of the zeolite. In these cases, enhanced sampling techniques need to be used to overcome these barriers and to simulate also these rare events. Hence, the enhanced sampling techniques distort time information, special techniques are required to obtain the MSD and thus the diffusion coefficients (see for example the work of Tiwary et al. [3]).
The proposed study should allow to achieve insight into the molecular factors governing the diffusion process. We should be able to derive the differences in diffusion between protic and aprotic molecules. Moreover the simulations should be able to reveal whether collective diffusion occurs, where various molecules travel collectively through the lattice. Such insights are truly innovative and have not been studied before.
This master thesis is on the cross roads between (non-equilibrium) thermodynamics, statistical physics and computational physics. On the one hand the student will need to understand the atomic and molecular physics involved in force field construction. On the other hand, statistical physics will be employed to understand the advanced sampling schemes. The topic aims at making the student comfortable with simulation techniques and statistical physics. If needed, the required programming skills will be transferred during the thesis.
The CMM has ample experience in modelling nanoporous materials, has developed over the years various methods to study phenomena at the nanoscale from statistical physics and from thermodynamics while relying on molecular input.
The proposed research is part of a larger concerted action in which physical and chemical transformations are studied at the nanoscale within nanoporous materials. In that framework the CMM collaborates with leading experimental and theoretical partners. It is the intention to involve the interested student in our work discussions with our collaborators.
Engineering & Physics aspects
Physics: use of quantum and classical mechanical models for materials modeling and thermodynamic properties.
Engineering: studying the MTO process for industrial purposes.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELLING, MATERIALS, NANOKeywordsmolecular simulations, Force fields, Free energy calculations, Nanoporous materialsReferences
[1]Efficient and Accurate Methods for Characterizing Effects of Framework Flexibility on Molecular Diffusion in Zeolites: CH4 Diffusion in Eight Member Ring Zeolites, Rohan V. Awati, Peter I. Ravikovitch, and David S. Sholl, The Journal of Physical Chemistry C 2013 117 (26), 13462-13473
[2] Molecular Mechanics Potential for Silica and Zeolite Catalysts Based on ab Initio Calculations. 2. Aluminosilicates, Joerg-R. Hill and Joachim Sauer, The Journal of Physical Chemistry 1995 99 (23), 9536-9550
[3] From Metadynamics to Dynamics, Pratyush Tiwary and Michele Parrinello, Physical Review Letters 2013 111, 230602
Contact
An Ghysels
Veronique Van Speybroeck