The development of advanced computer simulations to accurately predict the acidity of nanoporous materials
The development of advanced computer simulations to accurately predict the acidity of nanoporous materials
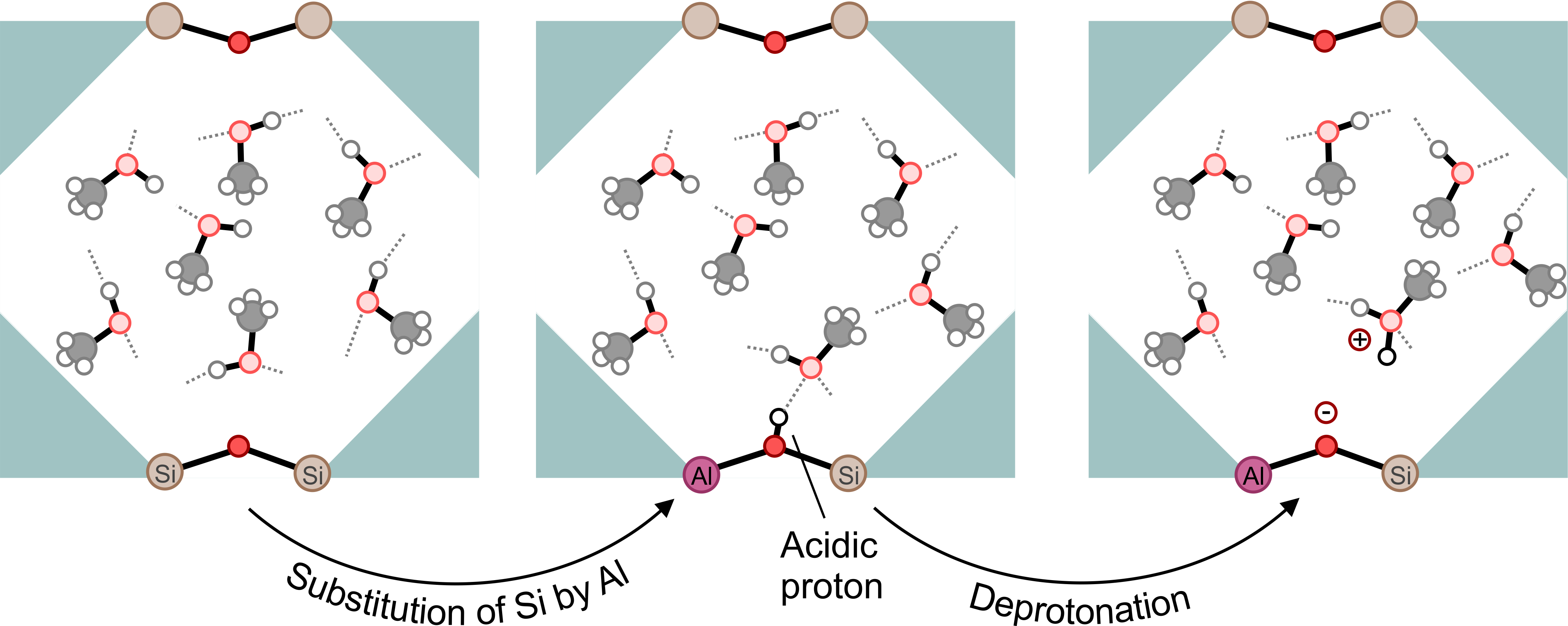
Promotor(en): V. Van Speybroeck /18NANO08 / Nanoporous materialsIn recent years, computational modeling has matured to a research field capable of reliably predicting the physicochemical properties of materials at a fraction of the time and economical expenses required to carry out the experiment. The Center for Molecular Modeling (CMM) has played a prominent role in the development of such advanced computational procedures, focusing on the accurate modeling of physical transformations and chemical reactions in a variety of nanoporous materials including zeolites and metal-organic frameworks. Zeolites, which are composed of corner-sharing SiO4 and AlO4 tetrahedra, are a class of microporous scaffold-like materials, making them attractive for a broad range of applications. The success story of these periodic materials can be largely traced back to their well-defined porous structure and their adjustable acidity, the latter determining the efficiency of the framework for a variety of applications. This acidity can be rationally engineered to a great extent through the gradual substitution of SiO4 by AlO4 tetrahedra, as the incorporation of aluminum in the framework also requires the inclusion of a cation – often a proton that binds to the oxygens in the zeolite framework – to preserve charge neutrality (see Figure 1). These acidic protons can later be donated by aluminum-substituted zeolites to the adsorbed guest species A, leading to the protonated AH^+ species (see Figure 1 for methanol as a guest molecule):
[zeo-H]+[A]⇌[zeo- ]+[AH+ ]
The efficiency with which this protonation of the guest species A takes place is determined by both the nature of the guest species and by the distribution and extent of aluminum sites and hence protons in the zeolite. To design a new generation of tailor-made zeolite materials, it is vital to obtain physical insight in how the aforementioned factors influence the acidity and can be rationally tuned from a microscopic point of view.
In principle, the computational modelling of protonated zeolites would allow for such a systematic investigation, by determining the free energy associated with the deprotonation. However, prior research at the CMM has highlighted two theoretical hurdles that still need to be overcome to reliably predict the acidity of zeolites. First of all, one needs to obtain an accurate estimate of the adsorption capacity of a given molecule in the pores of the zeolites at the elevated temperatures relevant for industrial applications. At these high temperatures, the adsorption capacity is expected to be influenced by the flexibility of the framework, as was observed for other microporous materials [1]. As a result, current methods to measure the adsorption capacity, which either rely on chemical intuition or assume the framework to be rigid, only yield approximate insight. Second, once the number of guest molecules in the framework is reliably predicted, the acidity of the zeolite needs to be determined. While several theoretical approaches exist, it is currently unclear to which extent they can be efficiently adopted to predict the acidity of zeolites [2].
Objective
The objective of the master thesis is to systematically investigate, from a physical point of view, how these two current challenges in the modelling of acidity may be overcome via the use of advanced molecular simulation methods. If successful, this thesis will result in the development of a first computational protocol to reliably predict the acidity of nanoporous materials for a variety of guest molecules.
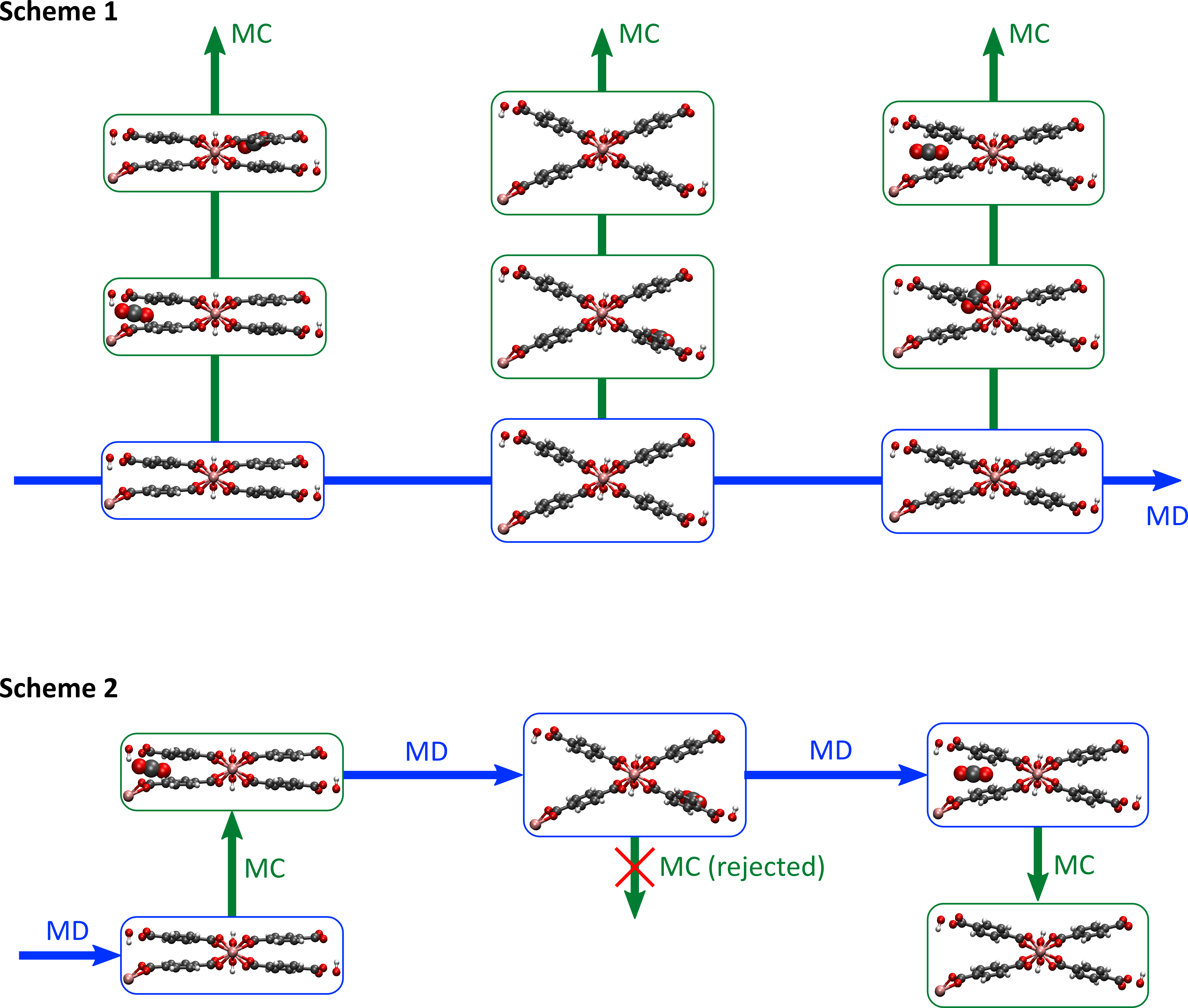
In a first step, we will focus on estimating the adsorption capacity of zeolites for a series of guest molecules at elevated temperatures. Traditionally, this is performed by classical Grand Canonical Monte Carlo (GCMC) simulations of the rigid framework. During this GCMC simulation, we try to add guest molecules to the host material (or remove them from the host material), something that will only succeed when the guest-host interactions are favorable. In this thesis, the result obtained with this traditional approach will be contrasted with two more advanced techniques (see Figure 2). In a first technique, a molecular dynamics (MD) simulation of an empty zeolite is performed for a sufficiently long time, and several snapshots of the zeolite are taken during this run. Afterwards, a separate GCMC simulation is started for each snapshot. When averaging over all these snapshots, which are assumed to be representative for the flexibility of the zeolite, the average amount of guest adsorption in the zeolite can be obtained [3]. In a second technique, recently developed at the CMM, a short MD simulation is initiated, after which we try to add or remove a guest molecule via an MC step. After this MC step, whether successful or not, we again perform an MD simulation, possibly with a different number of guest molecules. As such, we will obtain a sequence of alternating MD and MC steps, so that guest molecules can be removed or added during the hybrid simulation. The average amount of guest molecules is then again obtained by averaging over the whole MD/MC run.
In a second step, the acidity of the guest-loaded zeolite will be investigated. Hereto, two advanced computational schemes that were previously adopted to predict the acidity of dye molecules will be compared [2], and their results benchmarked against traditional approximate methods such as the adsorption enthalpy of ammonia. These advanced schemes are a prerequisite as the protonation of the guest molecule in a zeolite is a rare event, which is not sufficiently sampled in regular MD simulations. In a first advanced scheme, the free energy associated with the protonation is determined from a set of MD simulations during which the proton transfer from the zeolite to the guest molecule is carried out gradually. In a simulation, the position of the proton can be regulated via restrained MD, and the acidity can afterwards be determined from thermodynamic integration of the average forces acting on the proton in each of the restrained MD simulation. For this scheme to yield accurate acidities, however, is important to investigate how regulating the position of the proton influences the prediction. In a second scheme, called insertion/deletion, the protonation is divided into two separate steps, in which first the zeolite is deprotonated and afterwards the guest molecule is protonated. For both steps, the partial free energy difference can be determined from a set of advanced MD simulations in which a partial charge is associated with the proton. Afterwards, the total free energy of the protonation can be obtained by summing the two obtained partial free energy differences, from which the acidity of the zeolite can be predicted. For both schemes, it is important to properly take the dynamics of the acidic proton into account, as we have recently shown that the proton may participate in extended molecular configurations such as proton channels [3].
This master thesis, on the crossroads between statistical physics and computational material design, fits into a larger concerted action in which physical and chemical transformations in microporous materials are computationally investigated. As the CMM collaborates with leading national and international partners in the frame of this program, it is the intention to involve the interested student in our work discussions with our collaborators. The student will be made familiar with the simulation techniques necessary for this research proposal early on in the thesis, given the expertise available at the CMM.
Aspects
Physics aspect: Obtaining microscopic insight in the physical interactions between guest molecules and the host material governing the acidity of the material.
Engineering aspect: The optimization of zeolite design to promote their application for a variety of industrial applications.


- Study programmeMaster of Science in Engineering Physics [EMPHYS]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELINGKeywordsNanoporous materials, guest adsorption, acidity, Molecular dynamics, Monte Carlo, Free energyReferences
[1] M. Witman, S. Ling, S. Jawahery, P. G. Boyd, M. Haranczyk, B. Slater and B. Smit, "The Influence of Intrinsic Framework Flexibility on Adsorption in Nanoporous Materials," J. Am. Chem. Soc., vol. 139, no. 15, pp. 5547-5557, 2017.
[2] T. De Meyer, B. Ensing, S. M. J. Rogge, K. De Clerck, E. J. Meijer and V. Van Speybroeck, "Acidity Constant (pKa) Calculation of Large Solvated Dye Molecules: Evaluation of Two Advanced Molecular Dynamics Methods," ChemPhysChem, vol. 17, no. 21, pp. 3447-3459, 2016.
[3] C. Caratelli, J. Hajek, S. M. J. Rogge, S. Vandenbrande, E. J. Meijer, M. Waroquier and V. Van Speybroeck, "Influence of a Confined Methanol Solvent on the Reactivity of Active Sites in UiO-66," ChemPhysChem, vol. 19, no. 4, pp. 420-429, 2018.
Contact
Veronique Van Speybroeck