Defect migration in metal halide perovskites using machine learning potentials
Defect migration in metal halide perovskites using machine learning potentials
Promotor(en): V. Van Speybroeck /25657 / Nanoporous materialsBackground and problem
Perovskites are a broad class of materials which have the same type of crystal structure as CaTiO3, which was discovered in the early 20th century. They were named in honour of the Russian Count, Lev A. Perovski. Perovskites have ABX3 as chemical formula, with A and B cations and X an anion. There is a large and chemically diverse set of constructable perovskites, which can therefore be designed to exhibit a variety of exciting properties such as ferro-electricity (spontaneous polarization), magnetism, and superconductivity.
One particular class of perovskites, metal halide perovskites (MHPs), are now under world-wide investigation for an entirely different reason: they can be applied for photovoltaic (PV) purposes [1]. The most prominent MHP currently used is methylammonium (MA) lead iodide (MAPbI3), where the organic MA cation is encaged in a negatively charged lead iodide framework. In the last decade, the efficiency of perovskite-based solar cells has risen extraordinarily, from 3.8 % to 25.5 %, reaching an efficiency comparable to commercially produced silicon solar cells. Given their easier synthesis, direct band gap, lower cost, and tunable properties, MHPs have the potential to revolutionize the PV industry. While companies like Oxford PV are already investing to produce perovskite-based solar cells, MHPs still have stability issues. The most important concern is the low resistance of MHPs to moisture. Water easily penetrates the lead iodide cages and degrades the perovskite crystal structure. A possible solution is to use inorganic MHPs (e.g., replacing MA with Cs), although this destabilizes the wanted perovskite phase at room temperature to a metastable phase that will eventually transform to the stable non-perovskite phase with less interesting opto-electronic properties. At the center for molecular modeling (CMM), we recently collaborated with the Hofkens group in Leuven to develop a novel method for stabilizing the black phase, which was recently published in Science [2]. However, long term stability for CsPbI3 has not yet been achieved.
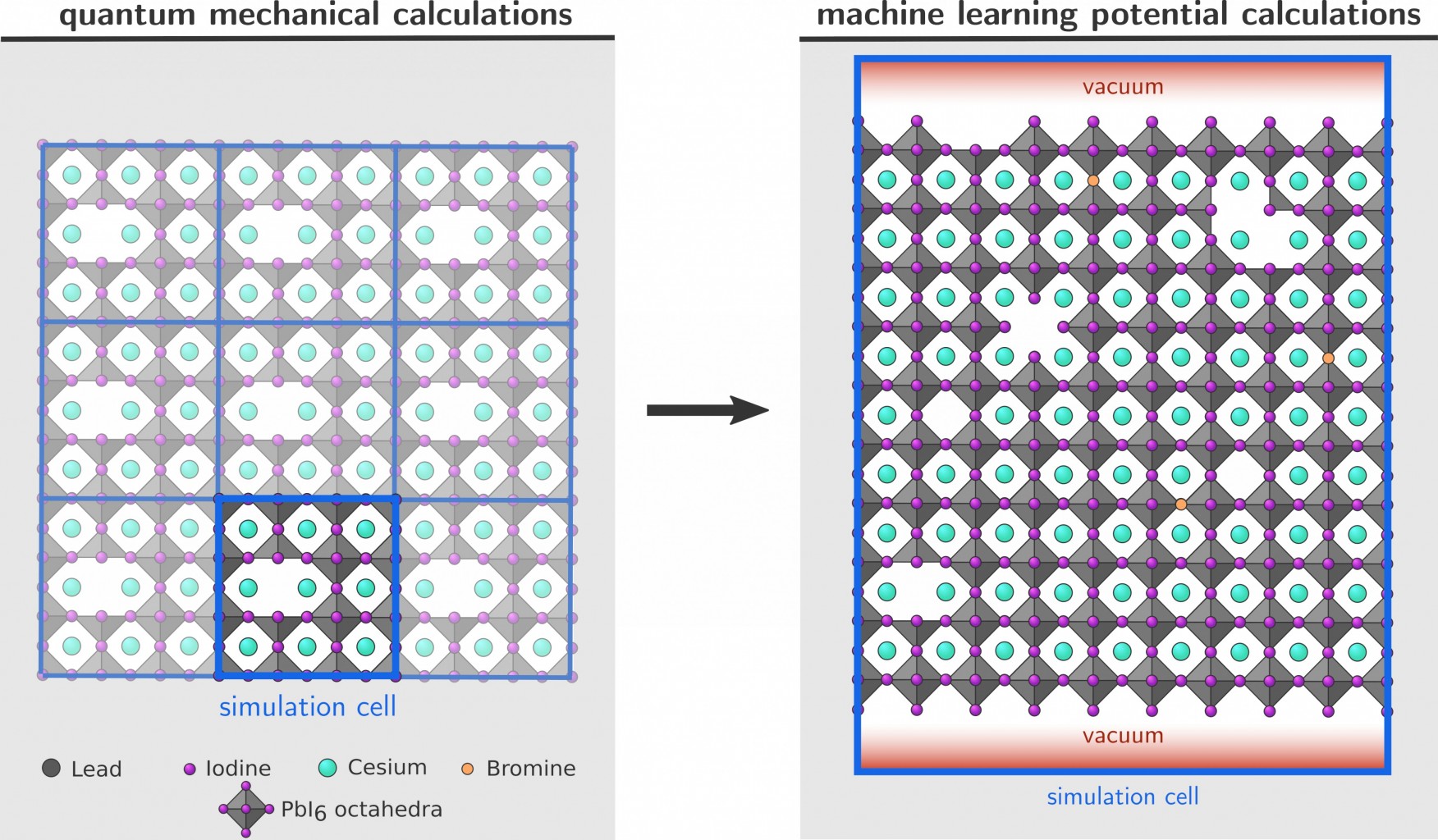
The perovskite community is actively searching for other (complementary) methods. Computational modeling is a valuable tool in this search for two reasons. On the one hand, it enables us to investigate the increased stability at the molecular level, learning new fundamental insights into the behavior of MHPs. On the other hand, we can easily screen the stabilizing methods for a large variety of MHPs, as it is easier to change the input settings for computational calculations than it is to redo experiments. Computationally, the material is modeled as an infinitely periodic structure with only a limited number of atoms in the unit cell, and its dynamic behavior may then be simulated using density functional theory (DFT) over a timescale on the order of 10ps (Figure 1, left). However, such an infinitely periodic structure is not a realistic representation of actual MHPs because it ignores intrinsic defects and the presence of crystal boundaries (Figure 1, right), which are nevertheless important for the stability of MHPs [3,4]. Defect migration, i.e. hopping of a defect from one site to another, is a heavily researched topic for MHPs as it plays an important role in the degeneration (and hysteresis) of MHP devices [5]. While it is possible to employ DFT when investigating e.g. the formation energy of isolated defects or the surface energy for a slab a few layers thick, more complex structures are unfeasible due to the much larger number of atoms in the unit cell. To overcome the computational limitations of DFT, it is possible to train a machine learning model to predict the potential energy surface of the system. Such machine learning potentials (MLPs) have already been developed successfully for a variety of systems.
Goal
The CMM has recently trained an MLP to describe CsPbI3 in the bulk (defect-free) phase. The goal in this thesis is then to further expand this MLP with the ability to model defects and surfaces 1. This will allow us to study the defect mobility in the bulk of the material, and understand how the mobility is influenced by the crystal boundaries. First, a new MLP that can describe defect and surface interactions has to be trained. The DFT data set that was used to train the MLP for bulk CsPbI3 is available, but additional DFT calculations will have to be performed in order to generate training data on various defect and interface structures. In the literature, there are already some examples on other materials [6,7]. Secondly, the new MLP has to be validated by comparing e.g. the defect formation and surface energy with DFT reference data. Afterwards, the MLP will be employed in large-scale simulations in order to study how various defects behave in bulk and near crystal boundaries. It will be interesting to see how well the MLP will perform when defects start to interact with each other; if necessary additional QM data will have to be generated.
As an example of what can be investigated, the new MLP can be used to study defect migration. The hopping of a defect from one site to another typically is a process that does not occur spontaneously, so advanced sampling techniques that push a defect towards another site will have to be applied to simulate the hopping. The CMM has ample expertise with these advanced sampling techniques to guide you with these simulations. Afterwards hopping rates can be determined for simulations with different concentrations of defects to see how this influences the hopping.
Experimental validation is possible based on TEM (transmission electron microscopy) or XRD (X-ray diffraction) experiments in collaboration with the EMAT group in Antwerp and the Hofkens group in Leuven.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsPerovskites, machine learning, photovoltaics, defects and surfaces, nanoscale modelingReferences
[1] W.-J. Yin et al., J. Mater. Chem. A 3, 8926-8942 (2015)
[2] Steele et al., Science 365, 679-684 (2019)
[3] Kye et al., J. Phys. Chem. 123, 9735-9744 (2019)
[4] Zhao et al., J. Am. Chem. Soc. 140, 11716-11725 (2018)
[5] Eames et al., Nat. Common. 6, 7497 (2015)
[6] Natarajan et al., J. Phys. Chem. C 121, 4368-4383 (2017)
[7] Byggmästar et al., Phys. Rev. B 100, 144105 (2019)
Contact
Veronique Van Speybroeck