Computational screening of drug storage in hybrid nanomaterials for drug delivery applications
Computational screening of drug storage in hybrid nanomaterials for drug delivery applications
Promotor(en): V. Van Speybroeck /25566 / Chemistry & BiochemistryBackground and problem
Drug development is one of the most strategic activities of pharmaceutical companies, with research and development (R&D) accounting for ca. 17% of their total capital expenditures in 2019. [1] Such high costs and a very low success rate (~4%) [2] of bringing new drugs into the market encourages the development of alternative strategies for drug formulations. One of them is to increase the efficacy of existing drugs by improving their access to the targeted tissues using drug carrier platforms.
There are numerous of these so-called drug delivery systems (DDS), traditionally divided in organic materials, with high drug affinity but uncontrollable drug release patterns, and inorganic materials, with extended porosity allowing controlled drug release but often displaying low drug loadings or toxicity. [3] Metal-organic frameworks (MOFs) have been suggested as a good combination of these two classes of materials. Due to the presence of both organic and inorganic moieties in their composition, they can yield the best characteristics of traditional drug delivery platforms.
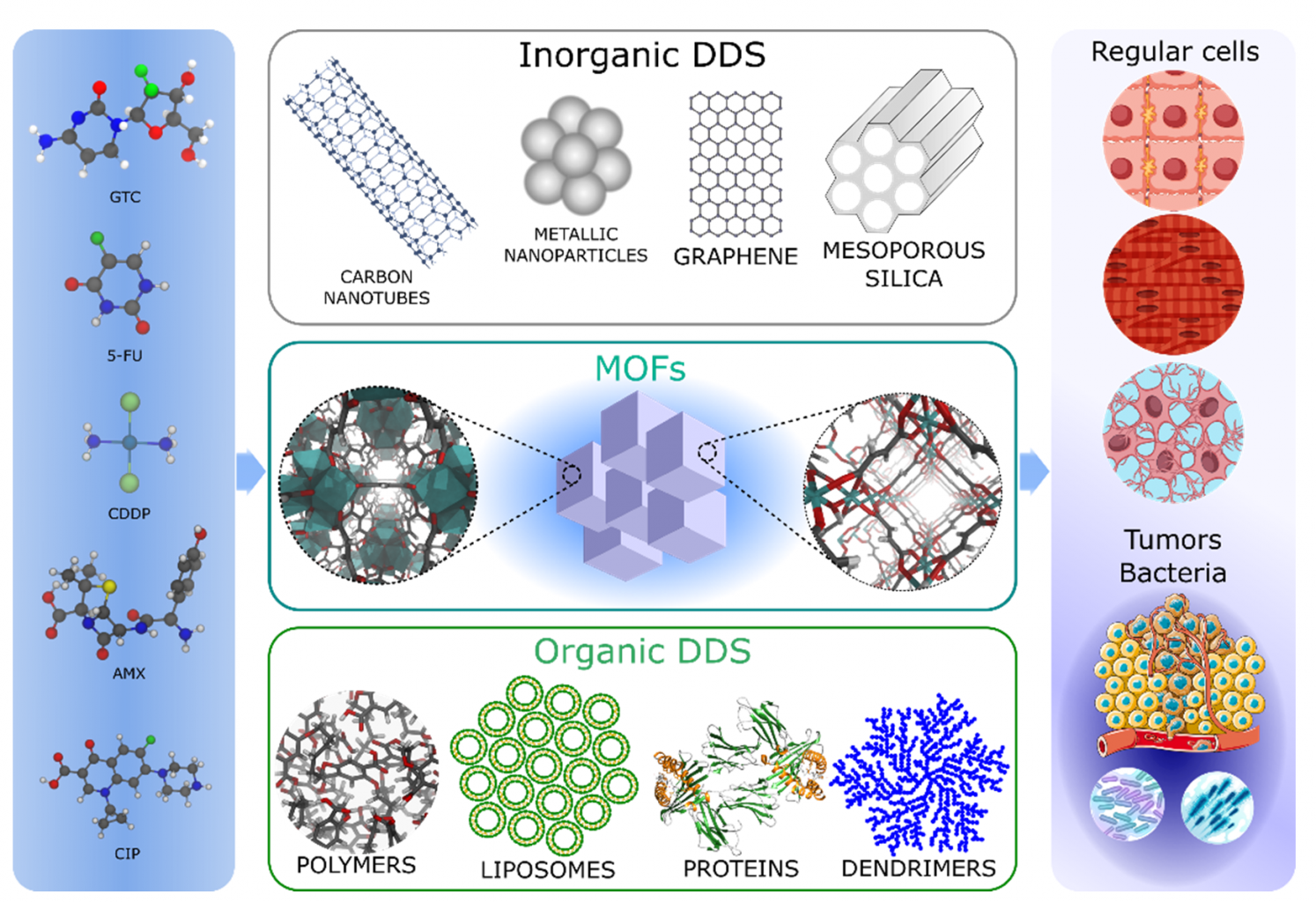
Figure 1. Scheme displaying different potential drug and MOF systems and the targeted delivery of drug systems to infected or tumoral tissues.
However, due to the large number of metal-organic frameworks and drug molecule candidates, listing good MOF/drug pairs has been a matter of serendipity so far. Although there is an increasing experimental and theoretical effort to characterize MOF-drug systems, [4] structure-property relationships allowing for a rational development of new and improved MOF materials for drug delivery applications are still lacking.
Goal
In this thesis, we propose an automated computational methodology to catalog the affinities of a large set of metal-organic frameworks [5] towards drug molecules with known solubility, stability and toxicity issues. [6] Such affinities will be determined by calculating the enthalpy of adsorption and the uptake of the drugs in each MOF using grand canonical Monte Carlo simulations integrated into an automated Python workflow. [7]
The maximum uptake obtained from these calculations will then be cross examined with respect to the textural characteristics (e.g. pore volume, surface area) and topologies of the initial MOF database. This will allow for a rationalization of the underlying structural parameters influencing the storage of different classes of drugs in MOF materials. An analogous protocol will be employed to determine structural features dictating the adsorption enthalpies of the drug entities in MOF scaffolds.
Finally, these results will allow to suggest systems with maximum drug uptakes and moderate adsorption enthalpies, parameters respectively correlated to optimal drug storage and controlled release. Such a library of optimal drug/MOF pairs will (i) from the basis of a training set for machine learning calculations devoted to develop even better drug-MOF systems, (ii) allow to further explore the relevant interactions in detail and scrutinize the optimal adsorption sites microscopically, and (iii) have their efficacy validated in vitro by an external collaborator.
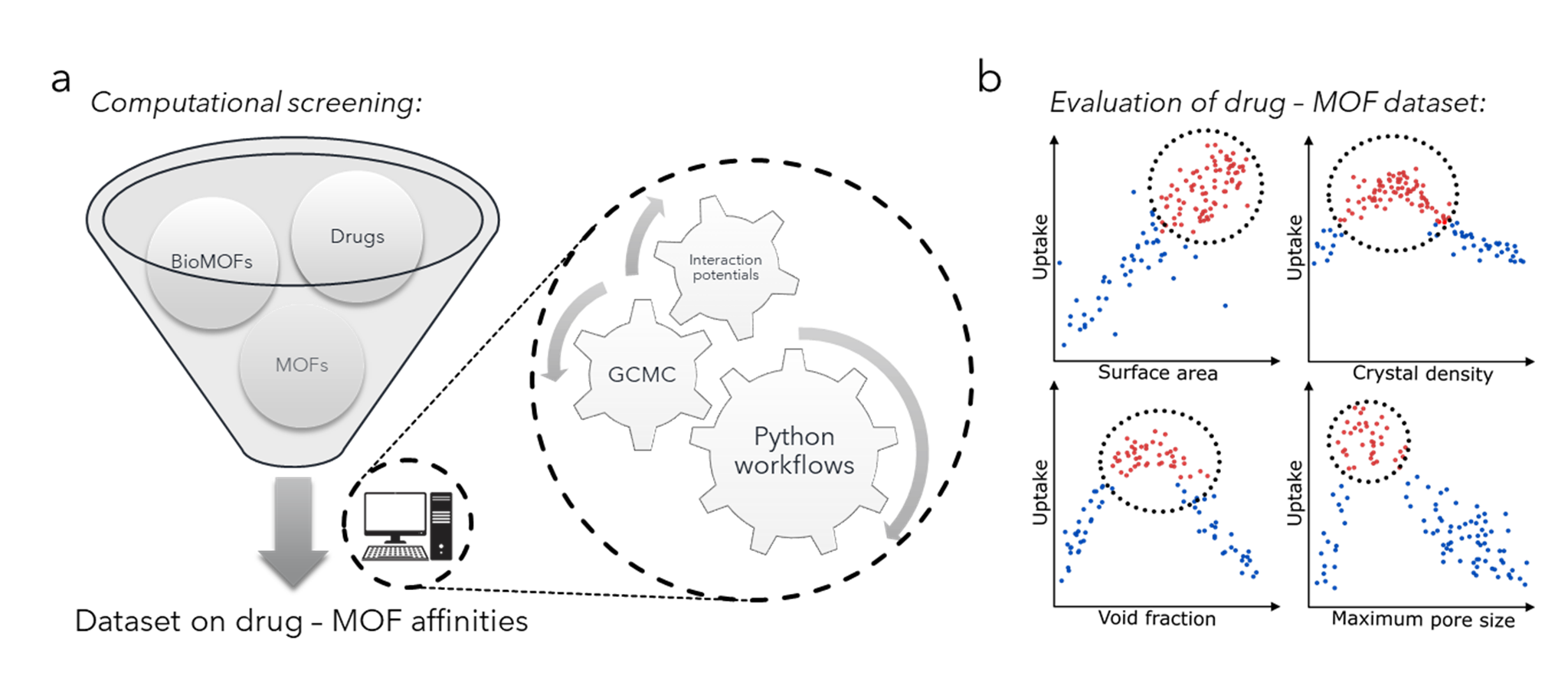
Figure 2. (a) Generation of a dataset of drug-MOF affinities from a computational screening of drugs and MOFs employing grand canonical Monte Carlo calculations automatized using Python workflows. (b) Promising drug-MOF pairs obtained from such screening will be compared to structural parameters from the MOF database to generate structure-property relationships allowing the design of new targeted materials for biomedical applications.
Within the scope of this work, the student will be coached early in the thesis in current Monte Carlo methods and in automated high-throughput Python workflows, allowing the acquisition of a highly-valued skillset to apply computational chemistry and data science for biomedical applications.
Remarks
Specialisation Biomedical Engineering: Biomechanics and Biomaterials
- Study programmeMaster of Science in Biomedical Engineering [EMBIEN]KeywordsH2 storage, gas displacement, clathratesReferences
1. Meagan Parrish Top 10 pharma companies by R&D spend Available online: https://www.pharmamanufacturing.com/articles/2020/top-10-pharma-companie... (accessed on Feb 8, 2021).
2. Hingorani, A.D.; Kuan, V.; Finan, C.; Kruger, F.A.; Gaulton, A.; Chopade, S.; Sofat, R.; MacAllister, R.J.; Overington, J.P.; Hemingway, H.; et al. Improving the odds of drug development success through human genomics: modelling study. Sci. Rep. 2019, 9, 18911, doi:10.1038/s41598-019-54849-w.
3. Mitragotri, S.; Stayton, P. Organic nanoparticles for drug delivery and imaging. MRS Bull. 2014, 39, 219–223, doi:10.1557/mrs.2014.11.
4. Mendes, R.F.; Figueira, F.; Leite, J.P.; Gales, L.; Almeida Paz, F.A. Metal–organic frameworks: a future toolbox for biomedicine? Chem. Soc. Rev. 2020, 49, 9121–9153, doi:10.1039/D0CS00883D.
5. Moghadam, P.Z.; Li, A.; Wiggin, S.B.; Tao, A.; Maloney, A.G.P.; Wood, P.A.; Ward, S.C.; Fairen-Jimenez, D. Development of a Cambridge Structural Database Subset: A Collection of Metal-Organic Frameworks for Past, Present, and Future. Chem. Mater. 2017, 29, 2618–2625, doi:10.1021/acs.chemmater.7b00441.
6. Gao, W.; Chen, Y.; Zhang, Y.; Zhang, Q.; Zhang, L. Nanoparticle-based local antimicrobial drug delivery. Adv. Drug Deliv. Rev. 2018, 127, 46–57, doi:10.1016/j.addr.2017.09.015.
7. Huber, S.P.; Zoupanos, S.; Uhrin, M.; Talirz, L.; Kahle, L.; Häuselmann, R.; Gresch, D.; Müller, T.; Yakutovich, A. V.; Andersen, C.W.; et al. AiiDA 1.0, a scalable computational infrastructure for automated reproducible workflows and data provenance. Sci. Data 2020, 7, 300, doi:10.1038/s41597-020-00638-4.
Contact
Veronique Van Speybroeck