Computation and partitioning of IR and Raman spectra to unlock the vibrational fingerprint of nanoporous adsorbents.
Computation and partitioning of IR and Raman spectra to unlock the vibrational fingerprint of nanoporous adsorbents.
Promotor(en): V. Van Speybroeck, L. Vanduyfhuys /20SPEC04 / SpectroscopyProbleemstelling:
Nanoporous materials, which are crystalline and have pores at the nanometer scale, have a wide range of applications ranging from gas separation and sensing to chemical catalysis. Zeolites provide an example of such materials and play an important role in the storage of thermal energy through the adsorption of water as well as in upcoming technologies to convert biomass or even CO2 into hydrocarbons, which are the building blocks for polymers. Given their economic impact, there is a powerful incentive for the characterization and smart design of new functionalized materials to obtain the best material for a given application. From an experimental point of view, it is a challenge to obtain insight into the microscopic structure of the material and even more when guest species are adsorbed inside the pores. However, recent advances made operando spectroscopic measurements possible. For example, infrared (IR) and Raman spectroscopy are especially well-suited to follow adsorbed molecules in the porous materials at operating conditions, as such allowing to generate a molecular fingerprint (as illustrated in the figure below). Unfortunately, it is extremely difficult to interpret the complex experimental spectra and it is in this respect that computational modeling offers a powerful complementary technique. Molecular simulations are able to accurately reproduce experimental spectra and allow to assign various spectral features to specific molecular motions. Unfortunately, this assignation is still a challenge in complex systems such as zeolites containing guest species, even with the help of theoretical techniques. On the one hand the full vibrational spectrum needs to be partitioned into separate vibrational modes that determine molecular motions involved in each mode, which becomes increasingly difficult with increasing system size. On the other hand the IR and Raman intensities need to be calculated for both the zeolite framework and the guest species requiring an expensive quantum mechanical description of the electronic structure.
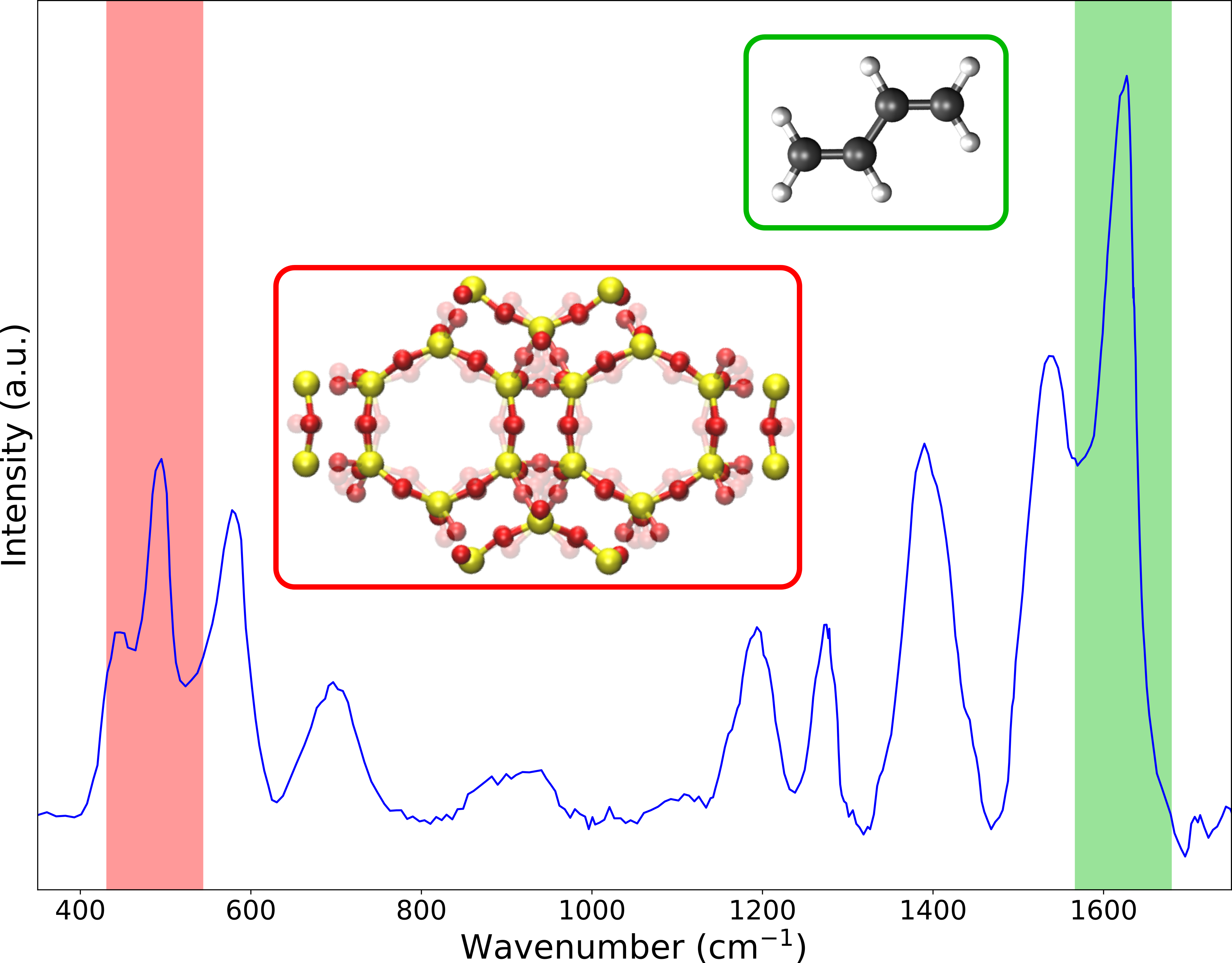
Doelstelling:
In this project the vibrational spectrum of zeolite frameworks hosting guest molecules will be calculated and analyzed in detail by means of computational modeling.
The first objective is to determine the vibrational modes of the host-guest system, for which two fundamentally different approaches exist: static or dynamic. The static approach starts from the equilibrium structure, which is a minimum on the potential energy surface (PES), and approximates the PES locally by means of harmonic functions, the so-called harmonic approximation. As a result, Newton’s equations of motion are solved analytically in terms of uncoupled harmonic oscillators at fixed frequencies. The corresponding normal modes represent the vibrational modes and this technique is labeled as normal mode analysis (NMA). However, finding the equilibrium configuration is not a trivial task. Furthermore, this static equilibrium does not account for the mobility of the guest molecules. Therefore, a dynamic approach, provided by molecular dynamics (MD), is required to sample the exact PES. Herein, Newton’s equations of motion are integrated numerically yielding a trajectory of the atomic positions as a function of time containing the sought-after vibrational modes. In literature, there already exist various procedures to extract such modes from an MD trajectory, such as Principal Component Analysis (PCA), which will be applied in this thesis. However, the student will be encouraged to propose extensions to NMA and PCA that promote more localized vibrational modes, allowing a more straightforward interpretation of the corresponding molecular motions.
A second objective focusses on computing the IR and Raman intensity corresponding with each vibrational mode. In case of NMA, each mode corresponds to a harmonic vibration at fixed frequency and hence corresponds with a single peak in the IR/Raman spectrum. The corresponding intensity can be computed from the fluctuating dipole moment and polarizability. In the case of PCA modes, one cannot necessarily assign each mode to a single frequency peak. However, one can still compute the corresponding intensity for each mode and compute its contribution to the full spectrum by means of a spectral decomposition.
As a final objective, the IR and Raman spectra of host-guest systems will also be computed directly as the Fourier transform of the time-dependent dipole moment and polarizability, respectively. The calculation of these quantities by means of ab initio methods has become possible since the development of the modern theory of polarization. These ab initio spectra can serve as a theoretical counterpart of the experimental spectra to assess the quality of the various computational models used throughout the thesis. Unfortunately, the dipole moment and polarizability are typically calculated for the complete unit cell, which does not allow for a straightforward assignation of each peak in the resulting spectra. Nevertheless, different localization techniques have emerged which could solve the problem, such as the use of Wannier centers. Although these techniques have proven their worth for the interpretation of vibrational spectra of solutions, they have never been used on nanoporous systems containing guest molecules. In this project, the performance of such localization schemes within zeolite systems will be assessed and applied.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsVibrational spectroscopy, normal mode analysis, infrared, Raman, quantum mechanical calculation, Molecular dynamics
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck