Is classic enough? Exploring how nuclear quantum effects affect proton mobility in zeolites through ab initio-derived machine learning potentials
Is classic enough? Exploring how nuclear quantum effects affect proton mobility in zeolites through ab initio-derived machine learning potentials
Promotor(en): V. Van Speybroeck /25618 / Nanoporous materialsBackground and problem
Zeolites are microporous aluminosilicate materials, with a pivotal role as catalysts in the chemical industry. They are composed of vertices-connected SiO4 tetrahedra and the flexibility of the Si—O—Si link gives rise to hundreds of possible different structures. In many of their most common catalytic applications, part of the Si4+ atoms in the zeolite framework are substituted by Al3+. This creates an excess of negative charge, that can be compensated by the addition of a proton (H+) on one of the oxygen atoms linked to the Al defect. Such a proton, as suggested by its commonly used name (Brønsted Acid Site or BAS in short), is strongly acidic and represents the archetypical active site for industrial zeolite applications such as olefin cracking, aromatic (de)alkylation, and the Methanol To Hydrocarbon (MTH) process [1].
Interestingly, the BAS is known for its dynamic nature at typical operando conditions [2]. Both theory and experiments have indeed shown that, in all key zeolite-catalyzed processes, organic substrates (such as aromatics, alkenes and even alkanes) exchange hydrogens with the framework already at temperatures as low as 298 K. Accurately modelling proton transfer reactions is therefore essential to obtain important fundamental insights into basically any relevant industrial process involving acidic zeolites, opening the path to a targeted improvement of the catalyst. Two effects are expected to play an important role while trying to compute a very accurate activation free energy for proton transfer reactions:
- A realistic modelling of entropic effects, which are relevant at the medium/high temperatures at which catalytic processes take place.
- An appropriate inclusion of the quantum nature of the BAS that, because of its low atomic mass, does not necessarily fit in the classical treatment normally reserved to nuclei.
While the use of advanced molecular dynamics techniques to solve the first problem is now a consolidated technique, the inclusion of Nuclear Quantum Effects (NQEs) in larger molecular systems still represents a relatively unexplored field of research as the computational power required for such calculations is prohibitively high using standard ab initio methodologies. In recent years, the use of machine learning has brought remarkable new possibilities to molecular modelling. By training a suitable model on ab initio-derived data, it is possible to obtain Machine Learning Potentials (MLPs) that reproduce the interatomic interactions with high accuracy at a reduced computational cost. Since NQEs are commonly introduced by coupling multiple regular molecular dynamics simulations in the Path Integral (PIMD) approach [3], ab initio data could be used to train a suitable MLP to describe the proton transfer process that, in its turn, would allow to perform the expensive PIMD simulations to recover the influence of nuclear quantum effects.
Goal
The goal of this thesis is therefore to obtain the most accurate barrier for proton transfer in the H-SSZ-13 zeolite thus far, including both entropic and nuclear quantum effects (Figure 1). Water, benzene, and simple alkanes and alkenes will be studied as proton transfer mediums, as they are representative for the most industrially relevant substrates. Initially, you will perform ab initio advanced sampling molecular dynamics to explore the process of interest, correctly accounting for entropic effects. Then, you will use that same ab initio data to train an MLP able to model the proton transfer with high accuracy and a dramatic reduction in the computational cost. Finally, you will employ the MLPs to recalculate the free energy barriers in the PIMD approach and evaluate how NQEs influence them at various temperatures.
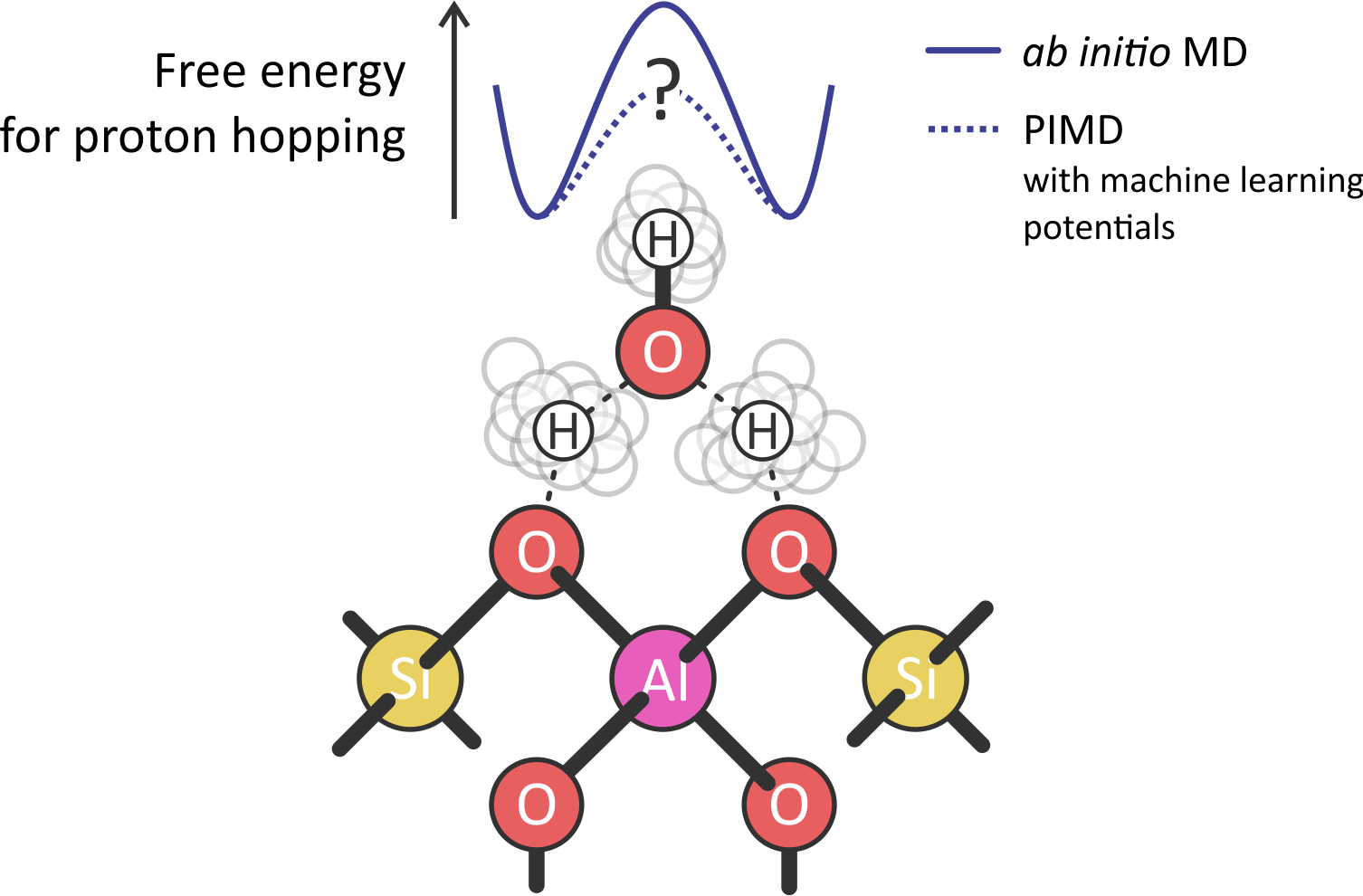
Figure 1: Schematic representation of the proton hopping between two oxygen atoms in a zeolite, mediated by a water molecule. The activation energy of the process (top) will change when going from a classical to a quantum description of the H nuclei.
The simplicity of proton transfer reactions represents an ideal case study for the development and assessment of this new methodology. Such an approach to tackle NQEs in realistic zeolite models has never been proposed before and represents a challenging, 360 degrees thesis subject. During the thesis period you will be able to work and get acquainted with three of the main state-of-the-art theoretical methodologies employed nowadays in molecular computational physics, namely advanced sampling ab initio molecular dynamics, machine learning derived force fields, and path integral molecular dynamics to introduce NQEs. The results you will obtain will be benchmarked against the large amount of experimental data available in the literature, usually derived from NMR spectroscopy by monitoring the H/D exchange on organic substrates (as an example, see Ref. [4]).
The Center for Molecular Modeling (CMM) has a large experience in the study of zeolites with advanced molecular simulations. You will be coached and guided to become acquainted with the software and techniques required to model and study the proton transfer process. The computational power necessary for the calculations will be ensured by CMM’s access to the main national supercomputing infrastructures.
- Study programmeMaster of Science in Engineering Physics [EMPHYS]KeywordsZeolites, machine learning, Molecular dynamics, Nuclear quantum effectsReferences
[1] Van Speybroeck, V. et al. Chem. Soc. Rev. 2015, 44, 7044-7111.
[2] Li, G. and Pidko, E. A. ChemCatChem 2018, 10, 1-24.
[3] Markland, T. E. and Ceriotti, M. Nat. Rev. Chem. 2018, 2, 0109.
[4] Chen, K. et al. J. Catal. 2017, 351, 130-135.
Contact
Veronique Van Speybroeck