Beyond periodicity: finite crystal modelling of metal-organic frameworks
Beyond periodicity: finite crystal modelling of metal-organic frameworks
Promotor(en): V. Van Speybroeck /28122 / Model and software development, Nanoporous materialsBackground and problem
Metal-organic frameworks (MOFs) are composed of inorganic and organic building blocks joined together with coordination bonds to form two- or three-dimensional extended network materials with fascinating and highly desirable physical properties. Noteworthy examples are MOFs exhibiting exceptionally high internal surface areas, ‘anomalous’ mechanical anisotropy such as auxeticity, negative gas adsorption, and the ability to undergo large-amplitude structural phase transitions induced by external triggers such as temperature or pressure. To enable the rational design of MOF materials for specific applications such as gas separation or nano-actuating devices, it is essential to establish structure-property relationships that elucidate framework behavior based on its structural description. Indeed, enormous research efforts have been dedicated to the development and application of novel computational and experimental methods which aim to understand and predict their physical properties. However, the complexity of these systems (as measured by the number of atoms of both the framework itself as well as its surrounding medium) is multiple orders of magnitude beyond the limits of even the most powerful heterogeneous (GPU/CPU) computing infrastructures.1 Most computational methods are forced to introduce various approximations and assumptions in order to make simulations tractable. One of these is the use of periodic boundary conditions to mimic the effect of bulk material in a relatively inexpensive manner (Figure 1). While effective, the assumption of infinite periodicity is highly unphysical and severely impairs the predictive capacity of current simulations. In reality, MOFs are inherently nonperiodic; they are finite in size (with diameters of 100 nm to 10 micron) and additionally exhibit a large array of nonperiodic phenomena that largely impact their physical properties.
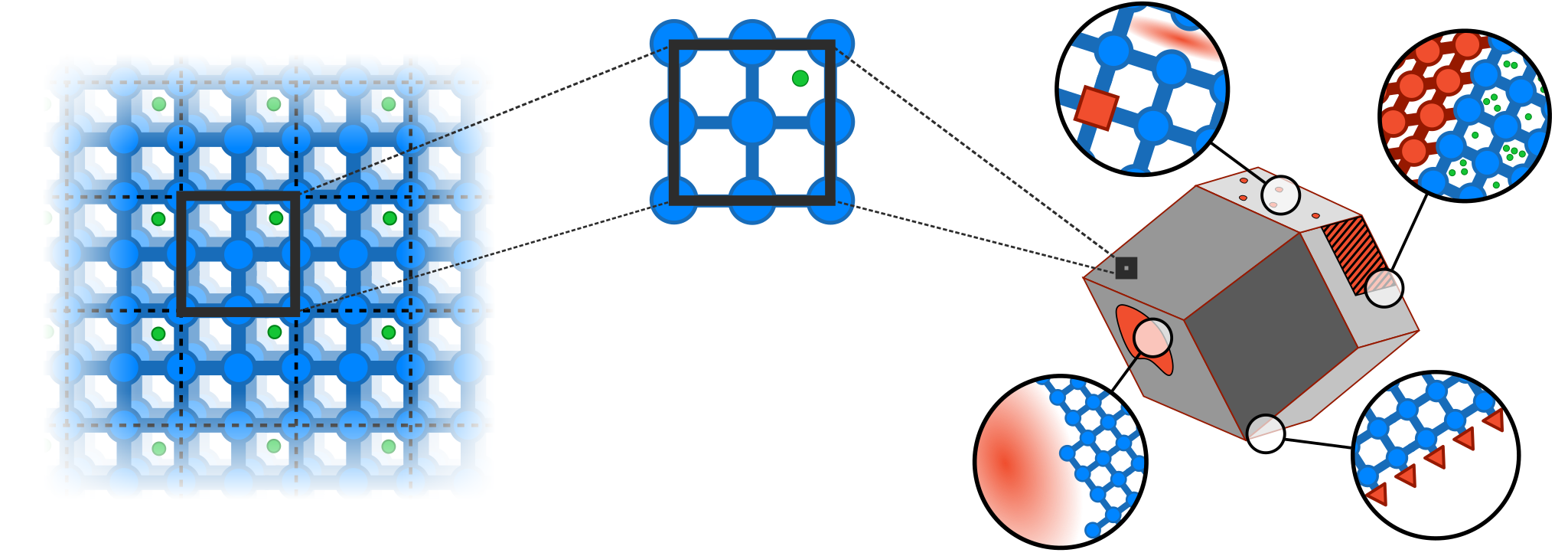
Figure 1: Schematic representation of framework materials, both in terms of a periodic abstraction used in computational models (left) as well as the realistic crystal observed in experiment (right).
While an atomistic treatment is typically the most accurate approach, it is often not strictly necessary to explicitly simulate each and every atom in the system. For example, global mechanical or thermal properties (bulk moduli, thermal expansion coefficients, pressure/temperature phase diagrams) often do not depend on the precise movement of each individual hydrogen in the system. This suggests that it is possible to consider so-called coarse-grained (CG) models in which irrelevant degrees of freedom are removed and only a small but important subset is retained. In physical terms, this means that several different atoms are no longer simulated separately, but are instead considered as a single group or bead (see Figure 2 for an example on UiO-66(Zr)).2 In machine learning terms, this is synonymous to reducing the dimensionality of the original atomistic phase space and marginalize the (Boltzmann) probability density. The main advantage of doing so is the fact that these models are orders of magnitude less expensive to simulate. Such approaches have therefore been extensively developed and successfully applied in large biomolecular systems.
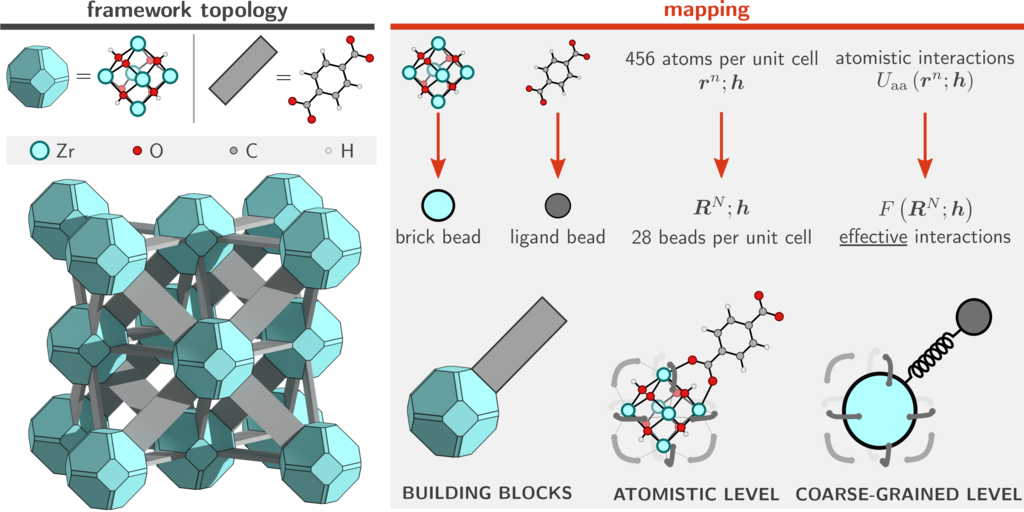
Figure 2: Coarse-grained model for the rigid UiO-66(Zr). The number of particles in the system reduces by more than an order of magnitude.
Goal
In this thesis, we aim to develop the first particle-based simulation technique for micrometer-sized MOF crystals. The target property of interest is the mechanical stability of the framework as a function of the size and morphology of the crystal as well as the pressure and temperature of the surrounding medium.3 We will start from existing CG models for bulk frameworks, and extend them towards finite crystals by deriving the effective interactions that occur near the surface of the crystal. Benchmark simulations of both atomistic and CG particles at a sub-micrometer scale will demonstrate the validity of the approach. Next, we will use experimental input about the average particle size and morphology to construct realistic structural models of MOF crystals. Finally, their mechanical properties are obtained based on massive simulations. Here, the student will have the opportunity to exploit the massive computing power of modern-day GPUs and perform the largest molecular dynamics simulations of these frameworks to date, with a target total system size of about 5-10 million atoms.4 The frameworks of interest are chosen based on their practical relevance and the availability of experimental data. In particular, we will investigate how the finite crystal size affects the structural flexibility of certain frameworks, for example in MIL-53(Al) or DMOF-1(Zn).
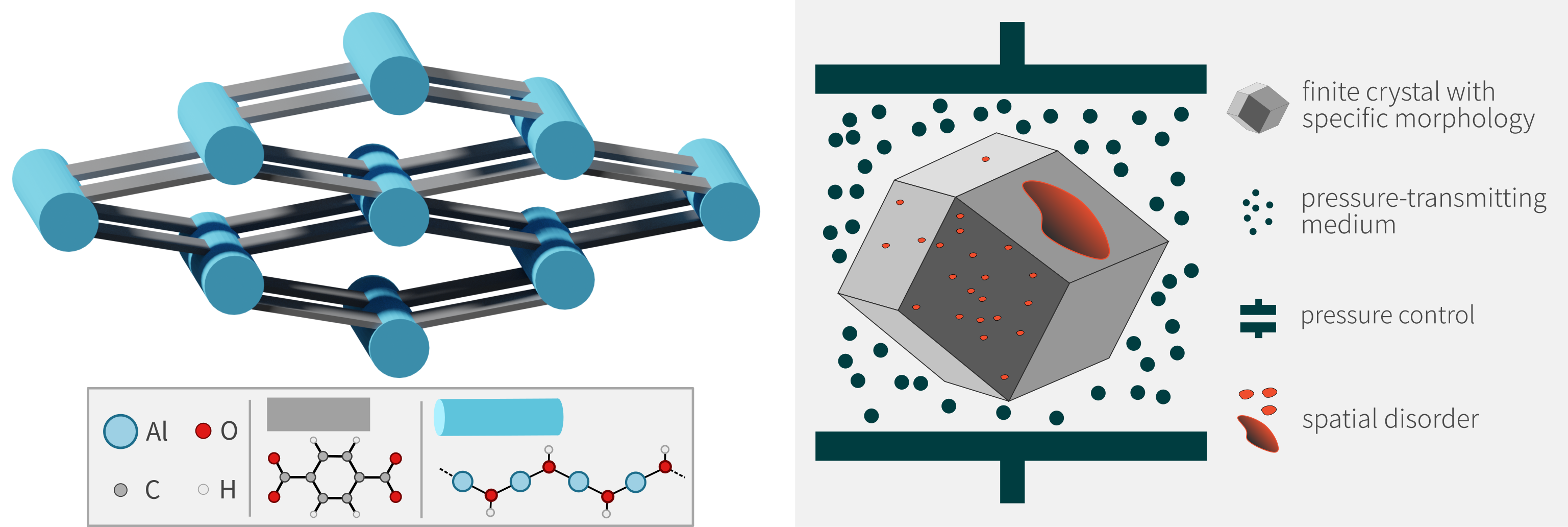
Figure 3: Overview of the flexible MIL-53(Al) framework (left) and the finite crystal methodology that will be developed in this thesis (right).
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsNanoporous materials, large-scale simulation, finite crystals, realistic materials, dimensionality reduction, coarse-grainingReferences
1V. Van Speybroeck, S. Vandenhaute, A.E.J. Hoffman, S.M.J. Rogge, Trends Chem. 3: 605, 2021.
2J.P. Dürholt, R. Galvelis, R. Schmid, Dalton Trans. 45: 4370, 2016.
3L. Schaper, J. Keupp, R. Schmid, Front. Chem. 9: 757680, 2021.
4S. Vandenhaute, S.M.J. Rogge, V. Van Speybroeck, Front. Chem. 9: 718920, 2021.
Contact
Veronique Van Speybroeck