Benchmarking systematic methods to identify appropriate collective variables in flexible metal-organic frameworks
Benchmarking systematic methods to identify appropriate collective variables in flexible metal-organic frameworks
Promotor(en): V. Van Speybroeck /18NANO11 / Model and software development, Nanoporous materialsNanoporous materials are omnipresent in a broad versatility of application domains such as energy storage, energy conversion, sustainable chemistry and biomedical applications. A special class of nanoporous materials are metal-organic frameworks (MOFs). These hybrid materials consist of metal-oxide clusters interconnected by means of organic linkers, resulting in a periodic framework with nanosized pores: a so-called nanoporous crystal. The discovery of MOFs therefore forms an important milestone in material science. Moreover, the enormous amount of linker/cluster combinations allows for tailor-made materials. Nevertheless, synthesizing all possible linker/cluster combinations would be an impossible endeavor. Model-guided design has therefore become indispensable in the development of new materials. At the Center for Molecular Modeling (CMM), computer simulations are applied to predict the thermodynamic behavior of MOFs and to guide the experimentalist towards desired applications.
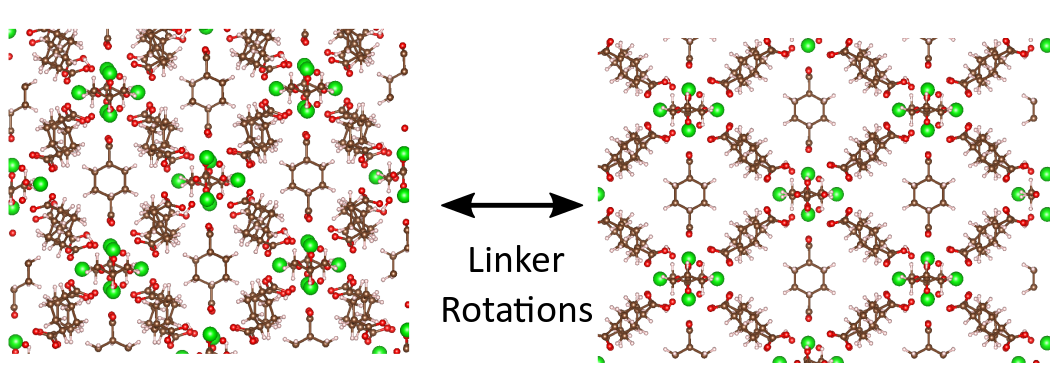
In this thesis topic we will focus on the inherent flexibility exhibited by an important subclass of these MOFs. Here, it is necessary to distinguish between the flexibility associated with physical transformations, such as the bending of linkers, and chemical transformations, such as the decoordination and recoordination of linkers in the structure. In both cases, the two (meta)stable states that define the begin and end state of the transformations are usually separated by large free energy barriers. As a result, these transformations, such as flexibility in MOFs, are rare events, posing three important challenges. First, an appropriate Hamiltonian describing the energy variations observed during a transition needs to be developed. For physical transformations, the detailed electronic structure can be neglected, while for chemical transformations an expense full quantum mechanical treatment is necessary. Second, the atomic motions which drive the flexible behavior in complex MOFs – the so-called collective variables – need to be identified. Third, since these transitions may occur on a long time scale, advanced simulations need to be performed. While the CMM has ample experience in these three steps [1], the appropriate set of collective variables for a given transformation (step 2) is often chosen in an ad hoc fashion. This master thesis focuses on the systematic identification of relevant coordinates to study rare events. This study will first be performed for physical transformations and, if possible within the time frame of the thesis, subsequent guidelines should be formulated to perform similar simulations for chemical transformations.
Objective
To study thermodynamic observables which are governed by long time scale events, such as the free energy difference between different (meta)stable phases, molecular dynamics simulations are often performed. However, as both the chemical and physical transitions under study are usually rare events, they are usually not sampled adequately in regular molecular dynamics simulations. Therefore, enhanced sampling techniques such as replica exchange and metadynamics are required. These techniques, which have already been implemented in our in-house developed molecular dynamics simulation package Yaff, are illustrated in Figure 2. In replica exchange, molecular dynamic simulations at different temperatures are performed in parallel. As activated events are preferentially sampled at high temperatures, endowing the material with sufficient energy to overcome the transition barrier, swapping between the various replicas in replica exchange promotes the sampling of rare events also at the lower temperatures of interest. While successful, this particular way of studying rare events is very inefficient, since the sampling of all degrees of freedom is enhanced simultaneously. In contrast, in metadynamics, solely certain slow degrees of freedom, the so-called collective variables, are enhanced, yielding a more efficient algorithm to study rare events. However, key to the application of these techniques is an adequate choice of the collective variables. Due to the large number of degrees of freedom involved in flexible MOFs, identifying those internal degrees of freedom that truly drive the transitions is far from a trivial task. During the master thesis, the student will work on identifying the collective variables solely based on the machine learning techniques. Recently, a large number of machine learning techniques, ranging from cluster methods to dimensional reduction methods, were proposed in the context of protein folding modeling [2]. Goal of this thesis is to apply similar techniques on framework breathing.
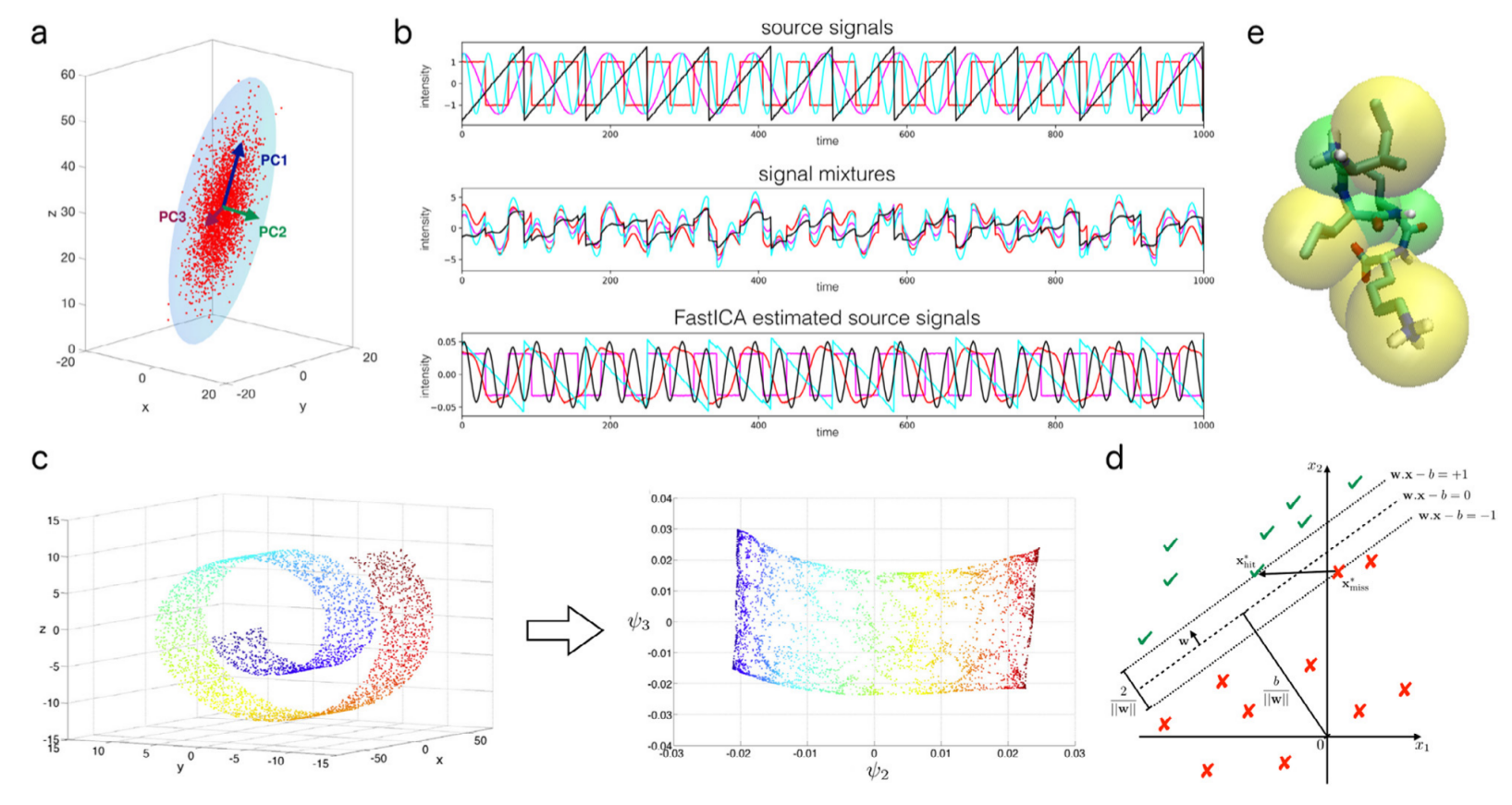
In a first stage, a large number of data on the flexible nature of the frameworks needs to be collected. To this end, replica exchange simulations will be performed. Based on this training data, the machine learning techniques can be unleashed. In the next step, the student will perform an extensive literature study to judiciously select those methods which are the most promising in the context of nanoporous materials and implement these methods in our in-house software packages. Based on his/her implementations, a benchmark study of the various methods needs to be performed, analogous to a previous free energy benchmark study performed at the CMM [1]. This benchmark study entails two aspects. First, a comparison needs to be made to investigate how the collective variables obtained with the several reduction techniques differ. Second, the precision and accuracy of the resulting free energy profiles in terms of these different collective variables need to be assessed. After identifying a good set of collective variables, an accurate free energy profile as a function of these collective variables can be constructed, yielding vital information on the flexibility of these MOFs. In a final step, guidelines should be proposed for the selection of collective variables based on machine learning techniques for the study of framework flexibility and their transferability towards other rare event studies. If possible within the time frame of this proposal, it would be worthwhile to investigate how the proposed methods perform in the context of chemical transformations, when accounting for the detailed electronic structure and including bond breaking.
The student will be actively coached to make him/her acquainted with the advanced simulations techniques early in the thesis year, and to transfer necessary programming skills needed to perform the research. Due to the challenging nature of the proposed topic, a strong motivation and a strong interest in programming is a must.
Engineering & Physics aspects
Physics: use of quantum and classical mechanical models for materials modeling
Engineering: engineering of materials for applications such as storage, separation, …


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELING, MATERIALS, NANOKeywordsmolecular simulations, Force fields, Free energy, Nanoporous materialsRecommended coursesSimulations and Modelling for the Nanoscale
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck