Band gap engineering by isovalent metal substitution in UiO-66
Band gap engineering by isovalent metal substitution in UiO-66
Promotor(en): V. Van Speybroeck /16SPEC03 / SpectroscopyMetal organic frameworks (MOFs) are porous, crystalline materials consisting of inorganic moieties (so called nodes) linked by organic ligands (linkers). The large variety of building blocks results in a high degree of tunability of the framework and makes these materials attractive for many applications. Their porosity paves the way for catalytic reactions since the materials offer a large and tunable surface area. In this thesis the UiO-66, a zirconium-based MOF, will be studied. UiO-66 is a promising material because of its high thermal and mechanical stability. These properties are of paramount importance for future industrial applications. The node of UiO-66 is an octahedron made of zirconium atoms. Each edge of this octahedron is connected with a linker, for example 1,4-benzenedicarboxilate, causing a coordination of 12 linkers per node (see figure). This highly coordinated configuration makes UiO-66 such a stable material.
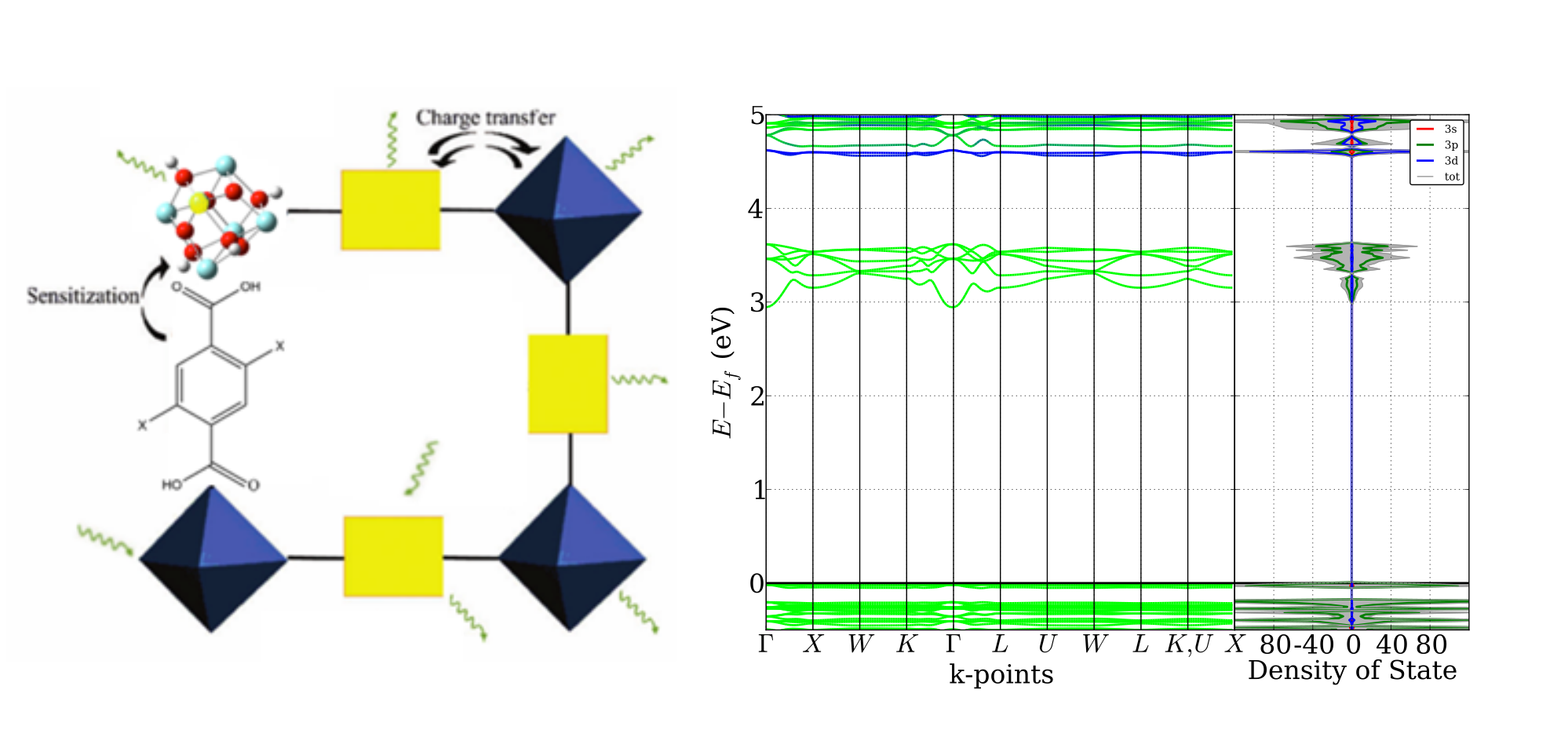
UiO-66 is a semiconductor, so its band gap (see figure) can be excited by light with a suitably chosen wavelength. For applications in photocatalysis, it would be interesting to be able to change the band gap. This effect was already shown in UiO-66 by incorporating functionalized linkers. However, the metal nodes can be altered as well. The effect of replacing zirconium by isovalent atoms (Ti, Hf), atoms with the same valence electronic structure, is still largely unknown territory. Investigating how the metals change the electronic properties, such as the band gap, the curvature of the conduction and valence band, and the spectroscopic properties, will enable us to gain more insight in how the composition of the node affects the materials properties and can lead to a completely new way of tailoring UiO-66.
Goal
In this thesis proposal, the electronic properties of UiO-66 will be studied by means of computational modeling. The goal of this thesis is to understand how changing the metals in the node affects the electronic properties of the framework. In particular, electronic properties will be studied by means of the VASP program, nowadays a standard code for periodic quantum mechanical calculations. The material will be constructed by substituting zirconium with isovalent atoms. One, two or even all zirconium atoms can be replaced by one type or a mixture of isovalent atoms. In this way, systematic investigation of the electronic properties will be possible. By calculating formation energies, you will moreover be able to evaluate the stability of all combinations. In addition to the periodic materials, also the separate building blocks will be studied: the node with the substituted isovalent metals and/or a cluster composed of a linker and a node. Considering these finite clusters, TD-DFT (Time-Dependent Density Functional Theory) calculations can be performed with another standard quantum mechanical code: Gaussian. TD-DFT allows you to calculate the excitation levels and to investigate the excited wavefunctions. This part of the thesis can be performed in parallel with the periodic calculations and will give complementary information on the electronic properties of the materials.
This thesis proposal is part of a concerted research action between the CMM and COMOC (Center for Ordered Materials, Organometallics and Catalysis, headed by Prof. Van Der Voort). As such the obtained data may be validated with experimental data.
Aspects
Physics: use of quantum mechanical models and solid-state concepts.
Engineering: application to the electronic properties of nanoporous materials.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELLING, MATERIALSRecommended coursesSimulations and Modeling for the NanoscaleReferences
K. Hendrickx et al., 'Understanding Intrinsic Light Absorption Properties of UiO-66 Frameworks: A Combined Theoretical and Experimental Study', Inorg. Chem. 54, 10701-10710 (2015). http://dx.doi.org/10.1021/acs.inorgchem.5b01593
L.-M. Yang et al., 'Computational exploration of newly synthesized zirconium metal-organic frameworks UiO-66, -67, -68 and analogues', J. Mater. Chem. C 2, 7111-7125 (2014). http://dx.doi.org/10.1039/c4tc00902a
Contact
Veronique Van Speybroeck