Applying advanced quantum mechanical techniques to describe d-orbital excitations in highly correlated systems
Applying advanced quantum mechanical techniques to describe d-orbital excitations in highly correlated systems
Promotor(en): V. Van Speybroeck /25250 / SpectroscopyBackground and problem
Metal-organic frameworks (MOFs) are a new class of porous materials built with metal-based units connected by organic linkers. These materials have unique properties and are very promising for numerous applications. However, the MOF formation process, i.e., the nucleation of isolated transition metal complexes (TMCs) to porous crystals, is not well understood. Spectroscopic techniques are an indispensable tool to understand this complicated process: when building blocks form larger units, the observed spectroscopic signals change and can be used to follow the nucleation in situ. UV-Vis spectroscopy can be employed to provide unprecedented insight in the characterization of the material as it sheds light on the electronic transitions in the system under study.
However, the experimentally observed spectra are often hard to interpret and it is complicated to relate observed signals to molecular structures of the involved complexes. To this end, computational methods are more and more used to determine the electronic structure and to predict and interpret the UV-Vis spectra.
An important spectroscopic fingerprint to follow during the nucleation process is the splitting of the d-orbitals of the transition metal when it coordinates with the ligands. Nevertheless, computationally characterizing the complicated electronic structure of these states is highly challenging. These correlated states cannot be written as a single Slaterdeterminant as they are prone to static electron correlation. High-level wave-function based ab initio methods beyond Hartree-Fock, such as complete active space self-consistent field (CASSCF) and multi-reference configuration interaction (MRCI), are therefore needed.
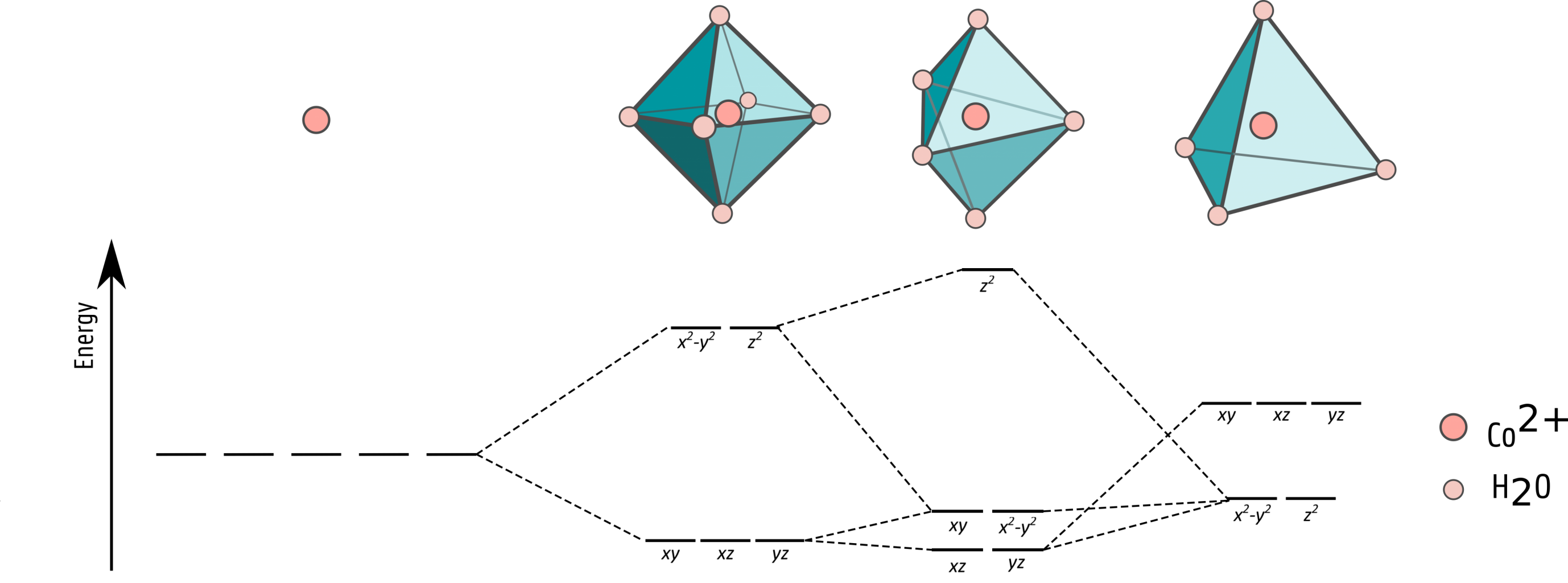
The calculation of excited state properties poses even more challenges. Transition energies and probabilities can, just like the ground-state properties, be calculated by wave-function based methods. However, applying these techniques to those complexes is not straightforward and computationally very expensive. Therefore one often relies on another many-body technique which is computationally much more attractive, namely time-dependent density-functional theory (TD-DFT). This technique is based on the one-dimensional density instead of on the much more complicated high-dimensional wave function. This is nowadays recognized as a powerful and reliable tool to reproduce excited-state properties for a wide range of molecular systems. Nonetheless, determining excited state properties of TMCs is not trivial with TD-DFT due to their complex electronic structure. LF-DFT on the other hand, which takes into account in a balanced way both dynamic and non-dynamic correlation effects, also seems to be a very promising technique for this task [3]. Therefore, a more thorough investigation on this matter is highly requested.
Goal
The objective of this thesis is to accurately describe d-d excitations in a series of d2–d8 hexaaqua coordinated TMCs [M(H2O)6]n+ with M a 3d transition metal ion (V2+/3+, Cr2+/3+, Mn2+/3+, Fe2+/3+, Co2+/3+, Ni2+/3+). These systems are highly interesting as they are the building blocks of MOFs. Moreover, they exhibit orbital occupation schemes ranging from 2 to 8 d-electrons from which we can gain new insights into the characteristics related to the number of d-electrons.
A first goal of the thesis is to systematically investigate the advantages and drawbacks of TD-DFT and LF-DFT in the study of the d–d transition energies and probabilities. The CMM has ample expertise with these techniques and the concepts of it are explained thoroughly in literature, for example in Refs. [1,2]. Furthermore, adequate experimental data are available and can be used to assess the accuracy.
Within this thesis, we will also investigate the influence of the ligand on the UV-Vis spectra. To this end, some of the water ligands in [M(H2O)6]2+/3+ will be replaced by ammonia or methanol resulting in TMCs with lower symmetry. This aspect is highly relevant for experiments as this symmetry reduction naturally takes place in the nucleation of TMCs in the formation of MOFs, but it has never been investigated before.
A thesis student choosing this topic should be interested in the fundamental quantum mechanical description of small molecular complexes and eager to apply various theoretical techniques to interesting complexes for which experimental data are available. The student will be actively coached to get acquainted as soon as possible with all necessary techniques to perform the suggested research successfully. The topic further builds on concepts such as many body techniques and spectroscopy which were taught within the course Atomic and Molecular Physics.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsSpectroscopy, time-dependent density functional theory, Quantum mechanics, transition metal complexes, symmetryReferences
List of figures: Figure 1: Splitting of the d-orbitals in TMCs with octahedral, trigonal bipyramidal, and tetrahedral symmetry.
References:
1. M.E. Casida and M. Huix-Rotllant, ‘Progress in Time-Dependent Density-Functional Theory’, Annu. Rev. Phys. Chem. 63, 287-323 (2012).
2. M. Atanasov and C. A. Daul and C. Rauzy, ‘New insights into the effects of covalency on the ligand field parameters: a DFT study’, Chem. Phys. Lett., 367, 737-746 (2003).
3. F. Vlahović et al., ‘Assessment of TD-DFT and LF-DFT for study of d-d transitions in first row transition metal hexaaqua complexes’, J. Chem. Phys. 142, 214111 (2015).
Contact
Veronique Van Speybroeck