Active Learning Neural Network Potentials for Hydrated Ionic Liquids
Active Learning Neural Network Potentials for Hydrated Ionic Liquids
Promotor(en): T. Verstraelen /28304 / Model and software developmentBackground and problem
Molecular dynamics simulations predict properties of condensed phases using only microscopic interactions as input. In an MD simulation, forces acting on the nuclei are used to solve Newton’s equation of motion, resulting in trajectories of atom positions through time. These forces are calculated using ab initio methods or so-called force fields. The former strategy relies on quantum mechanics to calculate the electronic structures and is usually very accurate, yet computationally prohibitive for systems containing more than a few hundred atoms. Alternatively, force fields link atom configurations to a system energy using a set of position-dependent functions that describe the interatomic interactions. Very often, the latter are fitted to ab initio data to obtain a quantitative model at affordable computational cost. Although many different force fields exist, few have been proposed that consistently describe chemical reactions.
In line with increasing interest in neural networks across many scientific fields, reactive neural network force fields are rapidly evolving.[1] While traditional force fields employ pre-imposed physical forms, neural network potentials rely on highly flexible, general functions capable of reproducing any potential energy surface. By training these neural network potentials on ab initio data, structure-energy relations of high accuracy can be obtained for complex chemical systems, affordable for subsequent molecular dynamics. Most successful are atomic neural network potentials which describe the total energy as a sum of atomic contributions, each calculated by a separate atomic neural network as a function of the local environment of the atom. Unfortunately, prediction errors of neural networks increase uncontrollably when extrapolating outside the training data, mandating data sets covering the entire phase space of interest. This is important as the behavior of a chemical system varies widely depending on concentration and conditions. As a result, it is difficult to maintain a balance between generating sufficient data and avoiding redundant, expensive calculations.
Active learning algorithms iteratively train models by identifying poorly represented regions of phase space and adding expensive reference data, specifically targeting these regions, for the next training iteration.[2] The identification of poorly represented regions occurs through test simulations using subsequent generations of the atomic neural network potential that is being trained on ab initio data. As such, the training data is gradually enriched as the test simulations generate new conformations.
In this MSc thesis, an automated active learning scheme (see Figure 1) will be developed and implemented for the training of atomic neural networks to reproduce density functional theory energies and forces. Hydrated NaOH will be used as a test case as it is a relatively simple system with well-known properties, facilitating model validation. It is also a prototype for more complex and technologically relevant (hydrated) ionic liquids, which are used in the development of new batteries, ultracapacitors for energy applications and in the synthesis of novel materials.
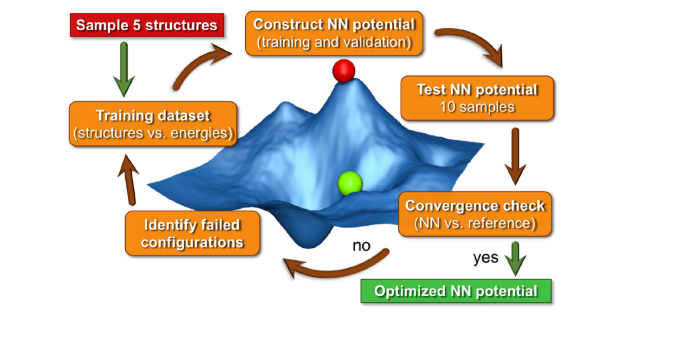
Figure 1: Workflow of an Active Learning algorithm [2]
Goal
Hellström et al. [3] have developed an atomic neural network potential for NaOH solutions, which can predict proton transfer and solvation effects. Their atomic neural network was trained on a database consisting of 16 113 structures with different NaOH concentrations using a combination of manual and random selection of concentrations, densities and system sizes. While it is possible, yet tedious, to make reasonable estimates of the ‘entire’ phase space for NaOH solutions, this becomes unfeasible for more complex ionic liquids. For example, important regions may be undersampled while in other regions, many redundant calculations may be included in the training set. To obtain sufficient data without wasting computational resources, an active learning algorithm will be developed, using the NaOH system as a test case, which can be compared to literature data. Practically, an open-source Python package will be developed, which should be general enough to allow later extensions to more complex phases.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]Keywordsmachine learning, Active Learning, Atomic Neural Network Potentials, Hydrated Ionic LiquidsReferences
[1] Behler, Angew. Chem. Int. Ed., 56, 1828 (2017)
[2] Loeffler et al., J. Phys. Chem. C, 124, 4907 (2020)
[3] Hellström et al., J. Phys. Chem. Lett., 7, 3302 (2016)
Contact
Toon Verstraelen