Accurate simulation of absorption spectra of acenes and metal-functionalized molecules using advanced electronic structure methods
Accurate simulation of absorption spectra of acenes and metal-functionalized molecules using advanced electronic structure methods
Promotor(en): K. Hemelsoet, V. Van Speybroeck /MM_14_SPEC_02 / SpectroscopySince the 1930s, the quantum theory of molecular electronic structure is known. For certain classes of molecules, typically when π-conjugation or transition metals come into play, conventional electronic structure methods are computationally too expensive. The active space of interest in such molecules exceeds 16 electrons in 16 orbitals, the current size limit for an exact solution. Quantum chemists then turn to approximative solution methods, which rely on a smaller active space or even a single reference Slater determinant. For excitation energies, two single reference methods are often used: equation-of-motion coupled cluster (EOM-CC) theory and time-dependent density functional theory (TD-DFT). Thus far these methods have been employed extensively, but without a thorough assessment of their accuracy for large active spaces.
In our group, a high-performance implementation of the density matrix renormalization group (DMRG) was recently written [1]. This method extends the size of active spaces for which an exact solution can be obtained to 40 electrons in 40 orbitals. DMRG hence provides the ability to assess the accuracy of EOM-CC and TD-DFT when the relevant active space is of intermediate size, far beyond the sizes which can be handled by conventional electronic structure methods.
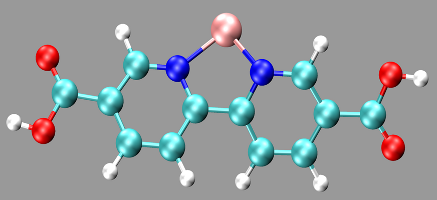
Goal Relatively simple molecules (polyenes and acenes) will be used as initial test system. The first objective of the master thesis is to calcualte excitation properties of these molecules and compare them with reference data available in literature. Various advanced electronic structure methods, in particular TD-DFT, EOM-CC and DMRG, will be applied. The aim of this study is to get acquainted with the various methods. In a next step, a more realistic systems will be examined. In particular, a metal-functionalized organic molecule (see the Figure) will be modeled and its excitation energy will be computed using the different methods. These functionalized molecules are important in modern research, since they form crucial building blocks of metal-organic frameworks (MOFs) with specific properties.
This master thesis combines a more theoretical/fundamental study and insight with practical simulations on realistic systems. The interested student will learn a variety of electronic structure methods. The needed software is available, and the interested student will need to get familiar with it. However, no programming is required. This topic contributes to current research activities at the Center for Molecular Modeling and intensive coaching will be provided. This challenging topic will make sure that the student learns a lot about various advanced electronic structure methods. Moreover, this insight will be applied on real systems and actual computations will be performed. Depending on the main interest of the student, the focus can lie on the theoretical part or on the applications.
Interests Second quantization, electronic structure methods
Interessante keuzevakken: Simulations and modeling for the nanoscale; VeeldeeltjestechniekenReferences
[1] S. Wouters, W. Poelmans, P. W. Ayers and D. Van Neck, CheMPS2: a free open-source spin-adapted implementation of the density matrix renormalization group for ab initio quantum chemistry, Computer Physics Communications, in press (2014), http://dx.doi.org/10.1016/j.cpc.2014.01.019 [ http://arxiv.org/abs/1312.2415 ].

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsElectronic structure methods, Spectroscopy, π-conjugation, Transition metals
Contact
Prof. Dr. ir. Karen Hemelsoet
Veronique Van Speybroeck