Accurate description of phase transitions in flexible materials using transferable machine learning potentials
Accurate description of phase transitions in flexible materials using transferable machine learning potentials
Promotor(en): V. Van Speybroeck, T. Verstraelen /25470 / Model and software developmentBackground and problem
Over the past three decades, metal-organic frameworks (MOFs) have emerged as an exceedingly promising class of nanoporous materials due to their extraordinary behavior and endless functional variety. They are composed of inorganic nodes and organic ligands, joined together with coordination bonds to form two- and three-dimensional networks. The vast number of possibilities in network topology and chemical composition of the organic and inorganic building blocks has given rise to a number of fascinating physical properties, such as their exceptionally high specific surface areas or the ability to undergo structural phase transitions induced by external stimuli such as breathing. These properties give MOFs a lot of potential for a wide variety of applications that involve either the separation or temporary storage of specific gases of interest such as methane, carbon dioxide and water. As a result, enormous experimental and computational research efforts have been dedicated to the design and synthesis of MOFs exhibiting targeted stimuli-responsive behavior for use in specific applications. Computational modeling at the molecular level is a particularly attractive approach as it allows to predict which MOFs are best suited for which applications without the need to perform a large number of expensive experiments. Naturally, such a computational approach requires highly accurate models that are able to describe the physics of these frameworks over a wide range of external conditions (pressure, temperature). Because of both the system size as well as the time scale of these phase transitions, it is computationally not feasible to tackle this problem using e.g. density functional theory (DFT). More approximate methods such as force fields have been developed in the past but they are considerably less accurate.
With the advent of large-scale GPU-accelerated computing infrastructure, more accurate approximations to the quantum mechanical interactions in molecular systems have been developed, using a variety of machine learning techniques [1, 2]. State of the art machine learning potentials (MLPs) have already demonstrated their immense capabilities on datasets of relatively small organic molecules, but have not yet been systematically developed for MOFs [3].
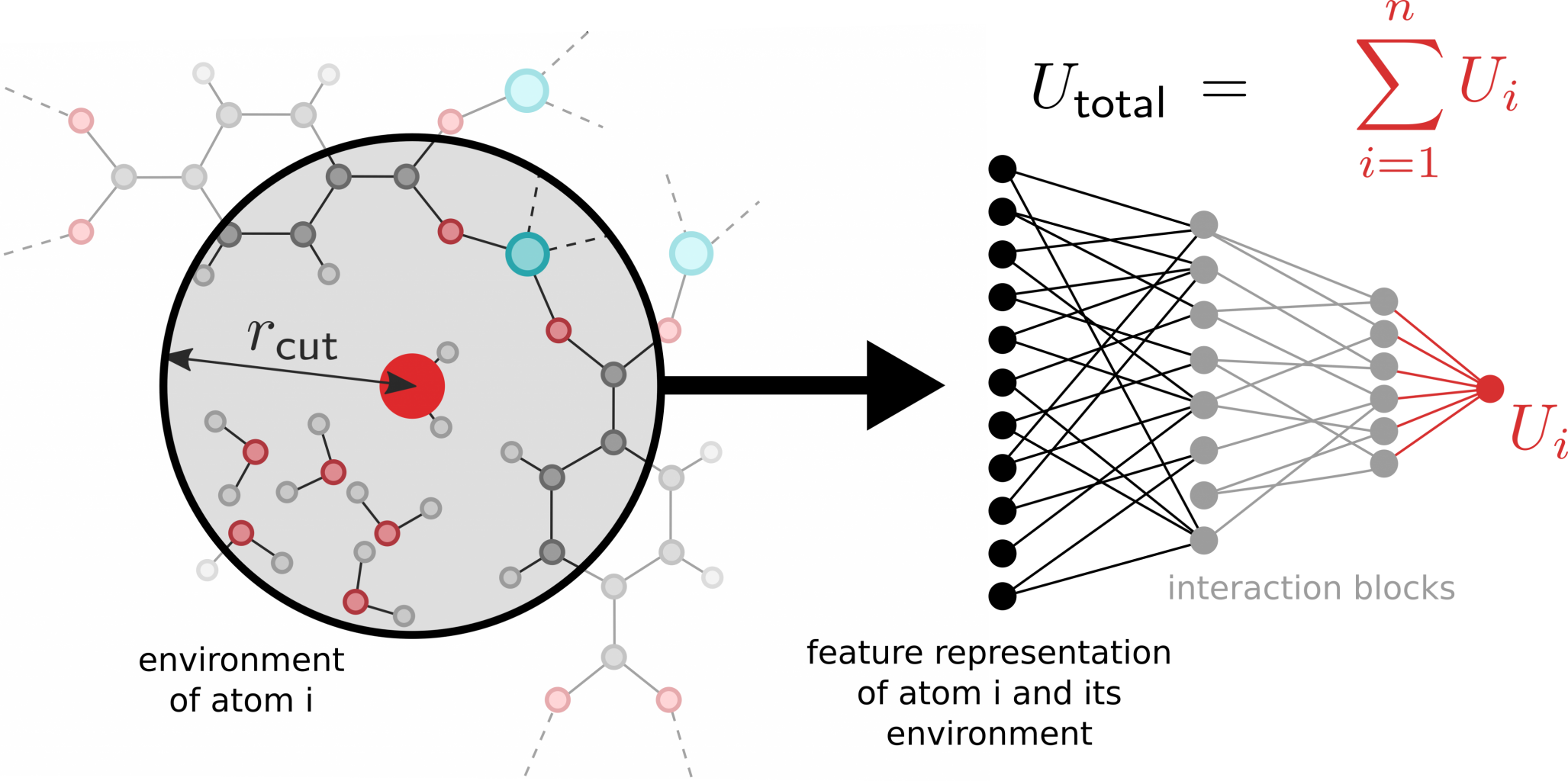
Goal
Here, we aim to develop a general methodology that efficiently constructs MLPs for flexible MOFs that are valid over a broad range of temperatures and pressures. The construction of MLPs essentially requires two main components. The first component is to obtain a large dataset of reference structures which are representative for the behavior of the material at a wide variety of external conditions (i.e. in all its metastable phases). An efficient and automatic sampling of such a dataset is far from trivial and will rely on enhanced sampling techniques in order to efficiently explore the phase space. Prior work at the CMM has already been performed in this area, and involved the use of free energy perturbation and normal mode sampling in order to avoid the computational cost of direct ab initio molecular dynamics (MD) simulations. The student is expected to further investigate how the data generation may proceed in a more efficient manner, and inspiration may be found in e.g. accelerated MD (aMD) or on-the-fly sampling schemes [4]. The second component consists of designing the internal architecture of the machine learning model. In recent years, many different architectures have been proposed in literature, but very few of them are suitable to describe the complex PES of flexible materials, in part because the number of atomic environments that the model has to learn can become much larger due to the existence of structurally different phases. While deep neural networks such as SchNet have demonstrated their ability to correctly predict forces in a wide variety of atomic environments, they are only able to achieve this once they have been trained extensively using massive amounts of quantum mechanical input data. However, such an approach may not be computationally feasible for the flexible materials in this thesis. Instead, the student will have to investigate ways in which the required amount of data may be reduced, for example by imposing certain physical constraints on the model (e.g. such that the long-range behavior between atoms resembles the classical electrostatic interaction).
To develop the methodology, we choose MIL-53(Al), a prototypical flexible MOF, as our benchmark system [5]. An initial dataset of quantum mechanical reference calculations is already available. First, several existing MLPs will be implemented and validated with their prediction of important physical quantities, such as (i) bond lengths and angles, (ii) pressure-versus-volume equation of state, and (iii) thermal expansion coefficients and vibrational frequency analysis. In addition, the student is encouraged to propose new models or dataset accumulation/augmentation schemes as mentioned earlier. After a rigorous validation of the developed model, it will be employed in large-scale simulations of the ‘breathing’ phase transition in MIL-53(Al) and others [6].
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]Keywordsmolecular simulations, Nanoporous materialsReferences
[1] K. T. Schütt et al. J. Chem. Phys. 148, 241722 (2018) https://doi.org/10.1063/1.5019779 [2] A. S. Christensen et al. J. Chem. Phys. 152, 044107 (2020) https://doi.org/10.1063/1.5126701 [3] M. Eckhoff et al. J. Chem. Theory Comput. 15, 3793-3809 (2019) https://doi.org/10.1021/acs.jctc.8b01288 [4] Donald Hamelberg et al. J. Chem. Phys. 120, 11919 (2004) https://doi.org/10.1063/1.1755656 [5] L. Vanduyfhuys et al. Nat. Commun. 9, 204 (2018) https://doi.org/10.1038/s41467-017-02666-y [6] J. Wieme et al. J. Mater. Chem. 7, 22663 (2019) https://doi.org/10.1039/C9TA01586H
Contact
Veronique Van Speybroeck
Toon Verstraelen