Ab initio studie naar de adsorptie en dimerisatie van C5-alkenen in Brønstedzure zeolieten
Ab initio studie naar de adsorptie en dimerisatie van C5-alkenen in Brønstedzure zeolieten
Promotor(en): V. Van Speybroeck, K. Hemelsoet /MM_14_NANO_12 / Nanoporous materialsAlkene conversions catalyzed by Brønsted-acid zeolites are of paramount importance for the industrial production of transportation fuels (gasoline, diesel, …) and base chemicals such as olefins and paraffins. Alkene cracking on H-ZSM-5 is particularly relevant for both classical FCC processes and emerging alternatives, e.g., the methanol-to-olefins (MTO) process. In both FCC and MTO processes, a considerable fraction of the primary products consists of heavier alkenes, and further cracking of these species is required to achieve high overall ethene and propene yields. Given the importance of these reactions, a profound understanding is required to select or design the best catalyst and to improve the performance of industrial FCC and MTO plants.
Adsorption of the reactants inside the pores of the zeolite catalyst is the starting point of any catalytic process. A detailed grasp of their adsorption behavior and accurate estimates of the corresponding adsorption enthalpy is therefore a prerequisite for the description of the complete catalytic cycles. However, depending on the nature of the alkene and the zeolite material, and external conditions such as temperature and alkene loading, physisorption and chemisorption occur in competition with side-reactions such as isomerization and dimerization, making it difficult to extract accurate data from experiments. Theoretical simulations can provide fundamental insights at the molecular level, and aid in understanding experimental observations.
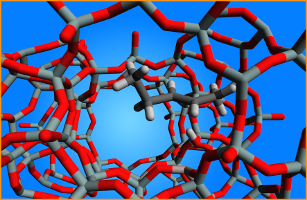
Figure 1. 2-pentene adsorbed at a Brønsted site in H-ZSM-5Objectives In this master thesis, the adsorption of C5-alkenes in H-ZSM-5 (Figure 1) and related materials will be investigated with advanced molecular modeling techniques. The goal is to identify the nature of the stable adsorbed states and intermediates at realistic temperatures and alkene loadings, and to determine accurate heats of adsorption by studying the contributions of the different zeolite-adsorbate interactions. State-of-the-art molecular dynamics simulations using periodic unit cells will be performed to account for conformational variations of the guest molecules and the flexibility of the zeolite lattice at finite temperatures. The Center for Molecular Modeling has a vast expertise in modeling nanoporous materials, and has access to the extensive computational resources required to study complex systems of scientific and industrial relevance. The research in this proposal will be conducted in collaboration with an experimental partner providing experimental input for setting up the simulations and validation of the obtained results. The proposed research work is diverse and challenging, and requires creativity and chemical intuition in addition to technical skills, which will be honed throughout the execution of the thesis.

- Study programmeMaster of Science in Chemical Engineering [EMCHEM]KeywordsHeterogeneous Catalysis, Chemical kinetics, Computational applications
Contact
Prof. Dr. ir. Karen Hemelsoet
Veronique Van Speybroeck
Kristof De Wispelaere