Ab initio force fields for flexible nanoporous materials
Ab initio force fields for flexible nanoporous materials
Promotor(en): T. Verstraelen, V. Van Speybroeck /MM_13_MODEV01 / Model and software development, Nanoporous materialsAbout 10 years ago, a new class of synthetic materials, metal-organic frameworks (MOFs), was discovered. These are crystalline materials consisting of metal ions or clusters connected by organic linkers to form a network of 1D, 2D or 3D channels with a diameter of a few nanometers. The main attractive property of these materials is the large internal surface area, going up to 5640 m²/gr for MOF-177. This means that one gram of material contains an internal surface as large as a football field. Hence enormous amounts of guest molecules can adsorb on this surface (cfr. a molecular sponge), leading to diverse industrial applications such as energy storage, CO2 capture, gas sensing, and so on. For certain MOFs, the selective adsorption of guest molecules is strongly influenced by the so-called breathing effect: the crystal framework changes its shape as function of temperature and loading. These exceptional properties have lead to a surge of interest in academia and industry, which is evident from the large number (>1000) MOF publications in highly-ranked journals.
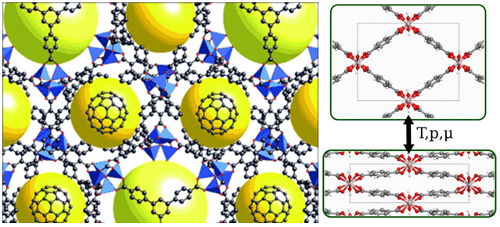
Figure: (left) an example of a MOF framework with cavities shown as yellow spheres Buky ball molecules are added for comparison. (right) Large- and narrow-pore structures of MIL-53(Al). Macroscopic properties of MOFs can be predicted with simulations of nanoscale models. This facilitates the systematic assessment of a large number of different (even hypothetical) MOFs for industrial applications: a first screening of the MOF database in search of the optimal material is performed in silico, prior to expensive wet-lab experiments. The two most common techniques are Molecular Dynamics (MD) and Monte Carlo (MC) simulations. For both methods, one relies on the Born-Oppenheimer approximation: atomic nuclei are treated as classical point particles on a potential energy surface (PES) governed by the surrounding electrons. In principle, the electronic structure must be solved with quantum mechanical methods (ab initio) to derive the potential energy felt by the nuclei. However, for simulations on MOFs, this becomes computationally infeasible and one has to use so-called force fields to compute the PES without explicitly describing the electronic structure. The main obstacle of a force-field simulation is the set of empirical parameters that must be determined. Historically, such parameters were determined from experimental reference data but today this is intractable due to the complexity of modern materials.
A new procedure was proposed at the Center for Molecular Modeling (CMM) for the development of a force field for the metal-organic framework MIL-53(Al) from ab initio reference data. The goal of the thesis is to apply this procedure to different variants of the MIL-47 framework. The shape and flexibility of the framework depends on the organic linkers, substituents on the linkers and the oxidation state of the Vanadium centers. In ab initio computations on single unit cells, the framework is more flexible compared to the experiment. With a force field model, it becomes possible to run geometry optimization and molecular dynamics simulations on supercells. These simulations can used to relate structural disorder (of the oxide chain, the linkers or the substituents) with the flexibility of the framework. This topic involves the development of new algorithms and theoretical models, and the implementation of these models in our in-house simulation software. If needed, programming skills will be transferred during the thesis.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsMetal-organic frameworks, Force fields, Atomic and molecular physics, Nanoscale, Molecular modeling, ProgrammingRecommended coursesComputational physics (C001827); Modeling and simulation at the nanoscale (E023370)
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck
Toon Verstraelen