Fast density matrix-based partitioning of the energy over the atoms in a molecule consistent with the hirshfeld-I partitioning of the electron density
Abstract
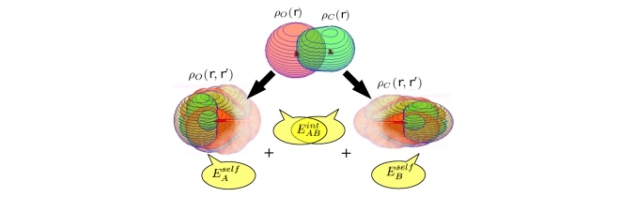
For the Hirshfeld-I atom in the molecule (AIM) model, associated single-atom energies and interaction energies at the Hartree–Fock level are efficiently determined in one-electron Hilbert space. In contrast to most other approaches, the energy terms are fully consistent with the partitioning of the underlying one-electron density matrix (1DM). Starting from the Hirshfeld-I AIM model for the electron density, the molecular 1DM is partitioned with a previously introduced double-atom scheme (Vanfleteren et al., J Chem Phys 2010, 132, 164111). Single-atom density matrices are constructed from the atomic and bond contributions of the double-atom scheme. As the Hartree–Fock energy can be expressed solely in terms of the 1DM, the partitioning of the latter over the AIM naturally leads to a corresponding partitioning of the Hartree–Fock energy. When the size of the molecule or the molecular basis set does not grow too large, the method shows considerable computational advantages compared with other approaches that require cumbersome numerical integration of the molecular energy integrals weighted by atomic weight functions. © 2011 Wiley Periodicals, Inc. J Comput Chem, 2011
 Open Access version available at
Open Access version available at