E. Van den Broeck
Stable Amorphous Solid Dispersion of Flubendazole with High Drug Loading via Solvent Electrospinning
Journal of controlled release
351, November 2022, Pages 123-126
2022
A1
Abstract
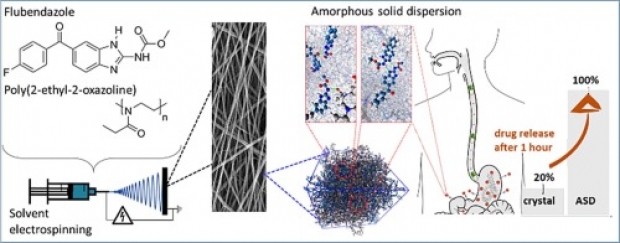
In this work, an important step is taken towards the bioavailability improvement of poorly water-soluble drugs, such as flubendazole (Flu), posing a challenge in the current development of many novel oral-administrable therapeutics. Solvent electrospinning of a solution of the drug and poly(2-ethyl-2-oxazoline) is demonstrated to be a viable strategy to produce stable nanofibrous amorphous solid dispersions (ASDs) with ultrahigh drug-loadings (up to 55 wt% Flu) and long-term stability (at least one year). Importantly, at such high drug loadings, the concentration of the polymer in the electrospinning solution has to be lowered below the concentration where it can be spun in absence of the drug as the interactions between the polymer and the drug result in increased solution viscosity. A combination of experimental analysis and molecular dynamics simulations revealed that this formulation strategy provides strong, dominant and highly stable hydrogen bonds between the polymer and the drug, which is crucial to obtain the high drug-loadings and to preserve the long-term amorphous character of the ASDs upon storage. In vitro drug release studies confirm the remarkable potential of this electrospinning formulation strategy by significantly increased drug solubility values and dissolution rates (respectively tripled and quadrupled compared to the crystalline drug), even after storing the formulation for one year.
A comparative theoretical study on the solvent dependency of anthocyanin extraction profiles
Journal of Molecular Liquids
351
2022
A1
Abstract
Anthocyanidins and anthocyanins are flavonoids with nutritional, antioxidative and color properties that are present in various food products and biomass, such as food waste. The large chemical diversity amongst these molecules potentially leads to different affinities or activities in food and non-food applications. In order to characterize the extraction profile, advanced analytical techniques along with optimized separation procedures are required. Alternatively, theoretical tools can be applied for predicting the solubility or binding affinity of molecules in various reaction media. In this paper, the solubility of anthocyanidins and anthocyanins was analyzed by various theoretical tools such as group contribution methods (e.g., Hansen solubility parameters and Flory-Huggins interaction parameter (χ12)) and molecular modeling (e.g., static calculations based on Density Functional Theory (DFT) and COSMO-RS). It was found that COSMO-RS was able to give quantitative information on the solubility behavior within various pure solvents and it is able to describe the main intermolecular interactions between colorant and solvent, while Hansen solubility parameters were most appropriate to find the most optimal organic solvent-water mixture ratio. In general, solvents with electron-rich aromatic rings and/or containing electron donors, acting as hydrogen bond acceptors, showed the highest solubilizing power for anthocyanidins and anthocyanins.
Gold Open Access
Unexpected formation of 2,2-dichloro-N-(chloromethyl)acetamides during attempted Staudinger 2,2-dichloro-β-lactam synthesis
European Journal of Organic Chemistry
2021, 42, 5823-5830
2021
A1
Abstract
In the quest for 3,3-dichloro-β-lactam building blocks, the serendipitous formation of 2,2-dichloro-N-(chloromethyl)acetamides was observed. This peculiar reactivity was investigated in detail, both experimentally and computationally by means of Density Functional Theory (DFT) calculations. 2,2-Dichloro-N-(chloromethyl)acetamides were thus shown to be formed experimentally through reaction of 2,2-dichloroacetyl chloride with glyceraldehyde-derived imines, i. e. (2,2-dimethyl-1,3-dioxolan-4-yl)methanimines, bearing aromatic N-substituents, in the presence as well as in the absence of a base. Deployment of aliphatic imines, however, resulted in complex reaction mixtures, pointing to the importance of a stabilizing aromatic substituent at nitrogen. The DFT results indicate that the substituents can alter the governing equilibria on the one hand and intrinsic barrier heights for the different routes on the other hand, showing that these are controlling the reaction outcome. Furthermore, the 2,2-dichloro-N-(chloromethyl)acetamides proved to be rather unstable in solution and thus difficult to isolate. Nonetheless, their molecular structure was confirmed by means of NMR analysis of several purified analogs and X-ray study of a 4-methoxyphenyl derivative.
Reductive imino-pinacol coupling reaction of halogenated aromatic imines and iminium ions catalyzed by precious metal catalysts using hydrogen
Journal of Catalysis
400, 103-113
2021
A1
Abstract
The first heterogeneously catalyzed process for the reductive coupling of imines and iminium ions is reported using precious metal catalysts in combination with hydrogen gas as the terminal reductant. The optimized method in terms of catalyst composition and reaction conditions allowed to produce aromatic vicinal diamines without the use of stoichiometric amounts of zero or low valent metals, which is currently the preferred method. The most important mechanistic features of the reaction were unraveled by a combined experimental and computational approach. The developed methodology is very efficient for the coupling of aromatic iminium ions with yields up to 88 % while imines give only low to moderate yields.
Gold Open Access
Access to bio-renewable and CO2-based polycarbonates from exovinylene cyclic carbonates
ACS Sustainable Chemistry & Engineering
9 (4), 1714–1728
2021
A1
Abstract
We investigate the scope of the organocatalyzed step-growth copolymerization of CO2-sourced exovinylene bicyclic carbonates with bio-based diols into polycarbonates. A series of regioregular poly(oxo-carbonate)s were prepared from sugar- (1,4-butanediol and isosorbide) or lignin-derived (1,4-benzenedimethanol and 1,4-cyclohexanediol) diols at 25 °C with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a catalyst, and their defect-free structure was confirmed by nuclear magnetic resonance spectroscopy studies. Their characterization by differential scanning calorimetry and wide-angle X-ray scattering showed that most of them were able to crystallize. When the polymerizations were carried out at 80 °C, some structural defects were introduced within the polycarbonate chains, which limited the polymer molar mass. Model reactions were carried out to understand the influence of the structure of alcohols, the temperature (25 or 80 °C), and the use of DBU on the rate of alcoholysis of the carbonate and on the product/linkage selectivity. A full mechanistic understanding was given by means of static- and dynamic-based density functional theory (DFT) calculations showing the determining role of DBU in the stability of intermediates, and its important role in the rate-determining steps is revealed. Furthermore, the origin of side reactions observed at 80 °C was discussed and rationalized by DFT modeling. As impressive diversified bio-based diols are accessible on a large scale and at low cost, this process of valorization of carbon dioxide gives new perspectives on the sustainable production of bioplastics under mild conditions.
Cation−π Interactions Accelerate the Living Cationic Ring-Opening Polymerization of Unsaturated 2-Alkyl-2-oxazolines
Macromolecules
53, 10, 3832-3846
2020
A1
Abstract
Cation–dipole interactions were previously shown to have a rate-enhancing effect on the cationic ring-opening polymerization (CROP) of 2-oxazolines bearing a side-chain ester functionality. In line with this, a similar rate enhancement—via intermolecular cation−π interactions—was anticipated to occur when π-bonds are introduced into the 2-oxazoline side-chains. Moreover, the incorporation of π-bonds allows for facile postfunctionalization of the resulting poly(2-oxazoline)s with double and triple bonds in the side-chains via various click reactions. Herein, a combined molecular modeling and experimental approach was used to study the CROP reaction rates of 2-oxazolines with side-chains having varying degrees of unsaturation and side-chain length. The presence of cation−π interactions and the influence of the degree of unsaturation were initially confirmed by means of regular molecular dynamics simulations on pentameric systems. Furthermore, a combination of enhanced molecular dynamics simulations, static calculations, and a thorough analysis of the noncovalent interactions was performed to unravel to what extent cation−π interactions alter the reaction kinetics. Additionally, the observed trends were confirmed also in the presence of acetonitrile as solvent, in which experimentally the polymerization is performed. Most intriguingly, we found only a limited effect on the intrinsic reaction kinetics of the CROP and a preorganization effect in the reactive complex region. The latter effect was established by the unsaturated side-chains and the cationic center through a complex interplay between cation−π, π–π, π–induced dipole, and cation–dipole interactions. These findings led us to propose a two-step mechanism comprised of an equilibration step and a CROP reaction step. The influence of the degree of unsaturation, through a preorganization effect, on the equilibration step was determined with the following trend for the polymerization rates: n-ButylOx < ButenOx < ButynOx ≥ PentynOx. The trend was experimentally confirmed by determining the polymerization rate constants.
 Open Access version available at UGent repository
Open Access version available at UGent repositoryGold Open Access
The potential of anthocyanins from blueberries as a natural dye for cotton: A combined experimental and theoretical study
Dyes and Pigments
176, 108180
2020
A1
Abstract
Natural dyes might be more environmentally sustainable compared to their synthetic counterparts, however in general their performance is worse. Therefore, typically metallic mordants are applied to improve the natural dye's affinity towards substrates, but this is not a suitable technique in a ‘green story’. In this paper, we test the potential of using anthocyanins from blueberry waste for dyeing cotton with biomordants, which are selected to tailor the intermolecular interactions such as hydrogen bonds, ionic bonds and π-π interactions with the dye molecule. In the experimental part, parameters during extraction and dyeing were optimized (e.g. temperature, pH, dyeing time and concentration). The effect of the (bio)mordants was monitored by Fourier transform infrared spectroscopy, spectrophotometric measurements and standard ISO wash and light tests. It was shown that stannous chloride stands out as metallic mordant, while no biomordants show sufficient intermolecular interactions to replace this metal salt. The experimental study has been corroborated with a series of molecular modeling calculations to obtain more insight into the intermolecular interactions between dye and (bio)mordants. To this end, both static Density Functional Theory based calculations as semi-empirical and force field based molecular dynamics calculations have been performed. The results indeed confirm that, in general, too small interaction energies for the biomordants of interest with the dye molecules are found, in correspondence with experimental findings. Overall, by performing systematic experiments in combination with the interpretation of the molecular models, this study yields valuable insights into the development of green routes towards use of anthocyanins as a natural dye for cellulose-based materials.
Open Access version available at UGent repositoryBrønsted Acid Catalyzed Tandem Defunctionalization of Biorenewable Ferulic acid and Derivates into Bio-catechol
Angewandte Chemie int. Ed.
59 (8), 3063-3068
2020
A1
Abstract
An efficient conversion of biorenewable ferulic acid into bio‐catechol has been developed. The transformation comprises two consecutive defunctionalizations of the substrate, that is, C−O (demethylation) and C−C (de‐2‐carboxyvinylation) bond cleavage, occurring in one step. The process only requires heating of ferulic acid with HCl (or H2SO4) as catalyst in pressurized hot water (250 °C, 50 bar N2). The versatility is shown on a variety of other (biorenewable) substrates yielding up to 84 % di‐ (catechol, resorcinol, hydroquinone) and trihydroxybenzenes (pyrogallol, hydroxyquinol), in most cases just requiring simple extraction as work‐up.
A switchable domino process for the construction of novel CO2‐sourced sulfur‐containing building blocks and polymers
Angewandte Chemie int. Ed.
58 (34), 11768-11773
2019
A1
Abstract
α‐Alkylidene cyclic carbonates (αCCs) recently emerged as attractive CO2‐sourced synthons for the construction of complex organic molecules. Herein, we report the transformation of αCCs into novel families of sulfur‐containing compounds by organocatalyzed chemoselective addition of thiols, following a domino process that is switched on/off depending on the desired product. The process is extremely fast and versatile in substrate scope, provides selectively linear thiocarbonates or elusive tetrasubstituted ethylene carbonates with high yields following a 100 % atom economy reaction, and valorizes CO2 as a renewable feedstock. It is also exploited to produce a large diversity of unprecedented functional polymers. It constitutes a robust platform for the design of new sulfur‐containing organic synthons and important families of polymers.