Towards a reliable quantum mechanical description of excited states in open-shell transition metal complexes
Towards a reliable quantum mechanical description of excited states in open-shell transition metal complexes
Promotor(en): V. Van Speybroeck /19SPEC03 / SpectroscopyElectronic transitions are at the basis of a wide range of atomic-scale phenomena. They are intimately linked to processes involving the absorption and emission of light studied in application domains such as spectroscopy, luminescence and photochemistry. Conversely, UV-Vis spectra shed light on these electronic transitions and are widely used within spectroscopy to characterize the structure of the material in which the transition takes place. Recently, UV-Vis spectroscopy was employed to investigate the formation of metal-organic frameworks (MOFs), which are a new class of porous materials built up from metal-based units connected by organic linkers. So far, however, the MOF formation process, from isolated transition metal complexes to porous crystals, is not well understood. In this respect, UV-Vis spectroscopy may provide unprecedented insight into the various steps of the nucleation process, as in transition metal complexes with partially filled d-orbitals, the so-called d–d transitions, fall into the UV, visible and near infrared region of the spectrum, and are hence uniquely traceable with UV-vis spectroscopy. When various building blocks form larger units, the observed signals change and these signals may thus be used to follow the nucleation process in-situ.
However, the experimentally observed spectra are often hard to interpret and it is often complex to relate observed signals to molecular structures of the involved complexes. To this end, computational methods are more and more used to determine the electronic structure of ground and excited states and to computationally predict the UV-Vis spectra. However, from a purely theoretical point of view, the accurate calculations of the electronic structure of ground and excited states of high spin complexes is very challenging. Within this thesis subject, we want to specifically investigate the accuracy of various electronic structure methods applied to various experimentally relevant high-spin transition metal complexes.
The electronic transition energies and probabilities can be calculated by high-level wave-function based ab initio methods beyond the Hartree-Fock level, such as the complete active space self-consistent field (CASSCF) and multi-reference configuration interaction (MRCI) methods. However, these methods are highly challenging and due to the expansion of the active space of the wavefunction, they are also computationally very expensive and only applicable to small model systems. To circumvent these problems, one often relies on another many-body technique which is computationally much more attractive, namely density-functional theory (DFT) and time-dependent DFT (TD-DFT) to calculate excited states. DFT is nowadays recognized as a powerful and reliable methodology to reproduce ground-state properties of molecules and even extended molecular systems, such as periodic MOFs. However, determining the wave function of high-spin complexes is not trivial with DFT and TD-DFT, as these states cannot be written as a single Slaterdeterminant and are hence prone to static electron correlation. Therefore, a more thorough investigation on this matter is highly requested, which is why this thesis subject focuses on the characterization of isolated transition metal complexes such as [M(H2O)6]2+/3+, which may be seen as building block for topical materials like MOFs. Adequate experimental data are available for such small high spin complexes, and can be used to assess the accuracy of the DFT and TD-DFT methodology.
Goal
The objective of the thesis is to investigate the performance of TD-DFT and other DFT-related methods to accurately reproduce the electronic d–d transitions in a series of d2–d8 hexaaqua coordinated transition metal ion complexes [M(H2O)6]n+ with M a 3d transition metal ion (V2+/3+, Cr2+/3+, Mn2+/3+, Fe2+/3+, Co2+/3+, Ni2+/3+). Furthermore, these results will be benchmarked with respect to more advanced wave-function based ab-initio methods.
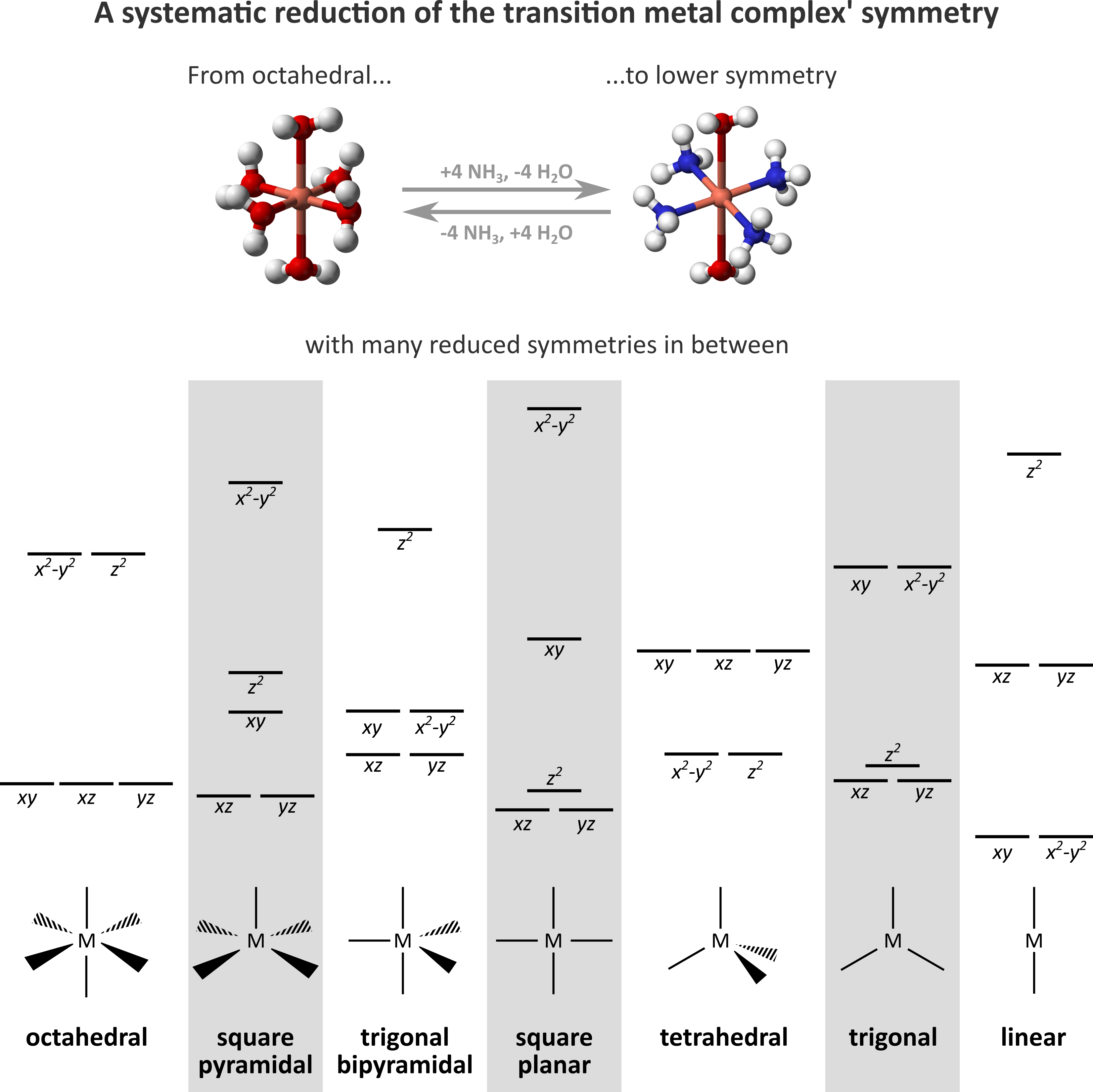
A first goal of this thesis is to perform an extended theoretical study to make the thesis student familiar with the different concepts of wave-function based methods such as CASSCF and MRCI. A good review on these accurate ab initio theoretical tools for the description of transition metal complexes is given in ref. [1]. One of the authors, F. Neese, is also main developer of the ORCA program package, one of the prominent codes in the field of advanced ab initio electronic structure methods. The concepts of TD-DFT are explained thoroughly in literature, for example in ref. [2], and in internal CMM notes.The second goal of the thesis is to systematically investigate the advantages and drawbacks of DFT-based methods in predicting the ground-state spin configuration of the different [M(H2O)6]2+/3+ complexes, and the d–d transition energies and probabilities. As these transition metal complexes are highly symmetric (see Fig.), the effective semi-empirical symmetry-based ligand field theory (LFT) seems a natural approach to interpret the extremely diverse excitation spectra and the transition probabilities, which following Fermi’s golden rule are related to the modulus squared of the matrix element of the dipole operator . While LFT also allows one to qualitatively determine the splitting pattern of the d-orbitals, as schematically shown, it fails for some high-spin complexes.
Within this thesis, we will investigate the influence of reduced symmetry on the predicted transition energies and probabilities by considering complexes in which the symmetry is systematically reduced, and compare to higher-level wave-function based methods. To this end, some of the water ligands in [M(H2O)6]2+/3+ will be replaced by ammonia or methanol resulting in transition metal complexes (X(H2O)n(NH3)6-n2+/3+) with lower symmetry. This aspect is highly relevant as the effects of reduced symmetry have never been investigated. Also when referring to the initial question of nucleation of transition metal complexes in the formation of MOFs, such symmetry reduction naturally takes place.
The thesis student choosing this topic should be interested in the fundamental quantum mechanical description of small molecular complexes and eager to apply various theoretical techniques to interesting complexes for which experimental data are available. The student will be actively coached to get acquainted as soon as possible with all necessary techniques to perform the suggested research successfully. The topic further builds on concepts such as many body techniques and spectroscopy which were taught within the course Atomic and Molecular Physics. Furthermore in this work, the student will gain insights in several symmetry concepts which are also introduced in the elective course “Symmetriegroepen”.
Aspects
Master of Science in Engineering Physics:This thesis subject is closely related to the following clusters: spectroscopy, model development, materials, nano, fundamentals. Physics aspect: use of quantum mechanical methods for spectroscopic modelling; Engineering: application to industrially relevant transition metal complexes and formation of nanoporous metal-organic frameworks.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) SPECTROSCOPY, NANO, MATERIALS, MODELLING, FUNDAMENTALSKeywordsSpectroscopy, time-dependent density-functional theory, Quantum mechanics, transition metal complexes, symmetryReferences
1. F. Neese et al.,’Advanced aspects of ab initio theoretical optical spectroscopy of transition metal complexes: Multiplets, spin-orbit coupling and resonance Raman intensities.’, Coord. Chem. Rev. 251, 288-327 (2007)
2. M.E. Casida and M. Huix-Rotllant, ‘Progress in Time-Dependent Density-Functional Theory’, Annu. Rev. Phys. Chem. 63, 287-323 (2012).
3. F. Vlahović et al., ‘Assessment of TD-DFT and LF-DFT for study of d-d transitions in first row transition metal hexaaqua complexes’, J. Chem. Phys. 142, 214111 (2015).
Contact
Veronique Van Speybroeck