Study of charge transfer in ionic liquids with constrained Density Functional Theory
Study of charge transfer in ionic liquids with constrained Density Functional Theory
Promotor(en): T. Verstraelen, D. Van Neck /17MODEV07 / Model and software development, Many-particle physicsIonic liquids (ILs) exclusively consist of cations and anions with strong Coulombic interactions, just like crystalline salts. In ILs, however, the Coulomb interactions are not strong enough to condense the ions into a crystalline framework. Instead, they form a liquid at a temperature below a (purely conventional) threshold of 100 °C. Still the Coulomb interactions are strong enough to cause several unusual properties not found in regular liquids, leading to various applications of ILs in industry.
Molecular simulation has always played an important role in IL research because some of their properties are difficult to understand from experiment alone. For example their heterogeneity, i.e. the structuring into microscopic polar and apolar domains, was first discovered in computer simulations and later confirmed by experiment.
Due to the large number of combinations of cations and anions, molecular simulations are valuable in the design of ILs for specific applications. However, the interactions between the ions are not merely Coulombic. They also transfer a small fraction of an electron and they polarize each other, leading to additional attractive forces that have a large many-body character. An accurate and efficient description of these many-body effects in molecular simulations of ionic liquids is still challenging.Goal
The goal of this thesis is (i) to compute the charge-transfer and polarization interactions between pairs of (oppositely charged) ions and (ii) to reproduce these results with approximate models that are applicable to ionic liquid models with many thousands of atoms.
The computations on the ion pairs will make use of the density-based energy decomposition analysis method. In this method, the charge of the ions is fixed by imposing constraints in the electronic structure calculation, i.e. no charge-transfer is allowed. The stabilization of the pair when this constraint is released is a good estimate of the charge-transfer interaction. The constrained DFT method will be implemented in this thesis and applied to pairs of ions from commonly used ionic liquids. This type of calculation is illustrated in Fig. 1, which shows the results for a constrained Hartree-Fock calculation. From a mathematical point of view, the constrained DFT method amounts to a non-linear optimization problem for the quantum-mechanical electronic wavefunction, where certain constraints are imposed on the solution.
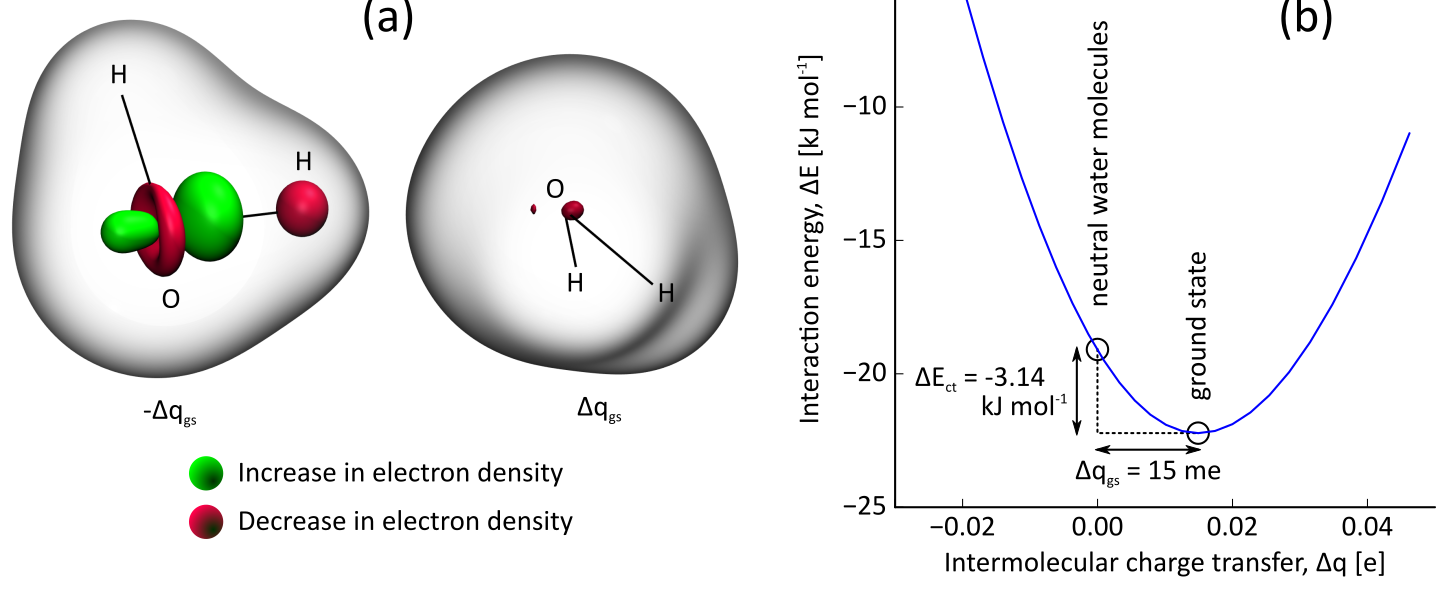
Fig. 1. Charge-transfer interaction in the water dimer. (a) Electron density deformation induced by the formation of a hydrogen bond between two water molecules. (b) Interaction energy of the water dimer as function of the amount of charge transfer.In a second stage, it will be investigated how these results can be reproduced by so-called electronegativity equalization methods. In such a method, the electronic energy of a molecule is approximated with a second order expansion in the atomic partial charges. Because this is a mathematically simple approach, it is easily applicable to systems with many thousands of atoms, e.g. the condensed phase of an ionic liquid. Recently, a new variant of this approach was developed at the Center for Molecular Modeling: atom-condensed Kohn-Sham DFT approximated to second order (ACKS2). Preliminary tests point out that this model is capable of describing noncovalent charge-transfer accurately. The results from the constrained DFT calculations, will be used to estimate parameters for the ACKS2 model.
In this thesis, the student will learn programming techniques to implement the constrained DFT method in the HORTON program. (An initial implementation will be provided, but it may need to be improved.) The student will also implement statistical methods to estimate model parameters in the ACKS2 model.Mobility: This project fits well in an ongoing collaboration with Mathieu Salanna (UPMC, Paris) who is an expert on the simulation of ionic liquids. The outcome of this thesis is directly relevant for his research and he can provide useful feedback during the thesis. A short visit to the UPMC, e.g. during the easter holidays, would be very valuable to analyse the results and to evaluate the impact of the findings on molecular dynamics simulations of ionic liquids.
Motivation Appl. Phys.: The implementation of the constrained DFT method and the development of a model for intermolecular charge-transfer require a fundamental understanding of the quantum-mechanical electronic structure of ion pairs. Such models will facilitate the design of ionic liquids, which a typical engineering problem.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELLING, FUNDAMENTALSKeywordsdensity-functional theory, charge transfer, ionic liquids, noncovalent many-body interactions
Contact
Dimitri Van Neck
Toon Verstraelen