Non-bonded forcefields from a density-based energy decomposition analysis: application to POSiSil materials
Non-bonded forcefields from a density-based energy decomposition analysis: application to POSiSil materials
Promotor(en): V. Van Speybroeck, T. Verstraelen /MM_14_MODEV_10 / Model and software developmentPOSiSils are a new type of materials currently in development at the COK (KU Leuven). These materials consist of silica building blocks connected by silicone linkers. The POSiSils share some similarities with zeolites and metal-organic frameworks, both nanoporous materials that receive a lot of attention in the scientific community. The most promising feature specific to POSisils is their flexibility combined with tunable hydrophobicity, rendering them ideal candidates for coatings of self-cleaning surfaces. However, the development of POSiSils is in an early stage and several steps still need to be taken in order to perfect the synthesis of these materials. This is why the Center for Molecular Modeling can offer support for our experimental partner COK (KU Leuven). By performing simulations on the atomic scale, a lot of insight in the properties of POSiSils can be gained, thus aiding the experimental development.
Some interesting features to simulate are vibrational spectra, flexibility and stacking configurations. However a lot of these simulations are very costly (if not impossible) at a quantum mechanical level. Classical molecular dynamics (MD) is a more attractive approach for atomic simulations that run over long length and time scales. In such simulations, the interactions between atoms are described by a force field. The accuracy of results obtained with these MD simulations depends crucially on the quality of the underlying force field. In principle, high-quality force fields can be derived from ab initio calculations such as Density Functional Theory (DFT). The main obstacle however, is that the ab initio energy has to be decomposed into parts that correspond to their force-field counterparts.
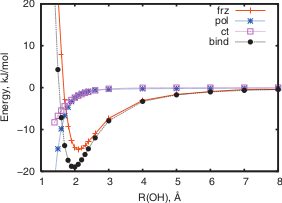
The goal of this thesis is to find force fields that accurately describe non-bonded interactions (such as electrostatics, van der Waals, ...) in POSiSil-like materials. We propose the following workflow. As a first step, ab initio reference data need to be obtained. This will involve a proper selection of different clusters and configurations and performing simulations at an appropriate level-of-theory. The second step aims at gaining a deeper understanding of the ab initio data. This can be done by performing a density-based energy decomposition analysis (http://dx.doi.org/10.1063/1.3253797). This novel method determines the energy decomposition from a variational principle, enabling a clean separation of different energy terms. In this step, also the influence of different density-partitioning schemes (Becke, Hirshfeld, ...) should be studied. Finally in the third and last step, the results from the second step will be used to derive non-bonded force fields that are tailored for POSiSil-like materials. In the end, this will provide deeper understanding of the interactions in POSiSils and lead to accurate simulations at large length and time scales of these exciting new materials.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]
Contact
Veronique Van Speybroeck
Toon Verstraelen