Using advanced molecular simulations to estimate the free energy of binding of glycans into proteins
Using advanced molecular simulations to estimate the free energy of binding of glycans into proteins
Promotor(en): A. R. Mehdipour, L. Vanduyfhuys /28180 / Chemistry & BiochemistryBackground and problem
Glycan-protein interactions represent an important class of biological interactions that occur between free or protein-attached glycans [1]. They are the basis for many prominent cell processes, including host-cell interactions during viral infection, such as with SARS-CoV-2 [2]. In a lot of biological processes, interaction between the glycan and the partner protein strengthens the overall interaction between two interacting proteins [3] (Figure 1). Given the importance of glycan-protein interactions, there is ongoing research dedicated to understanding the role of these interactions. However, estimation of the glycan-protein binding affinity is a hard task because of several factors. First, protein-attached glycans are remarkably flexible. Second, glycan-protein interactions are generally weak, therefore demanding highly sensitive estimation methods.
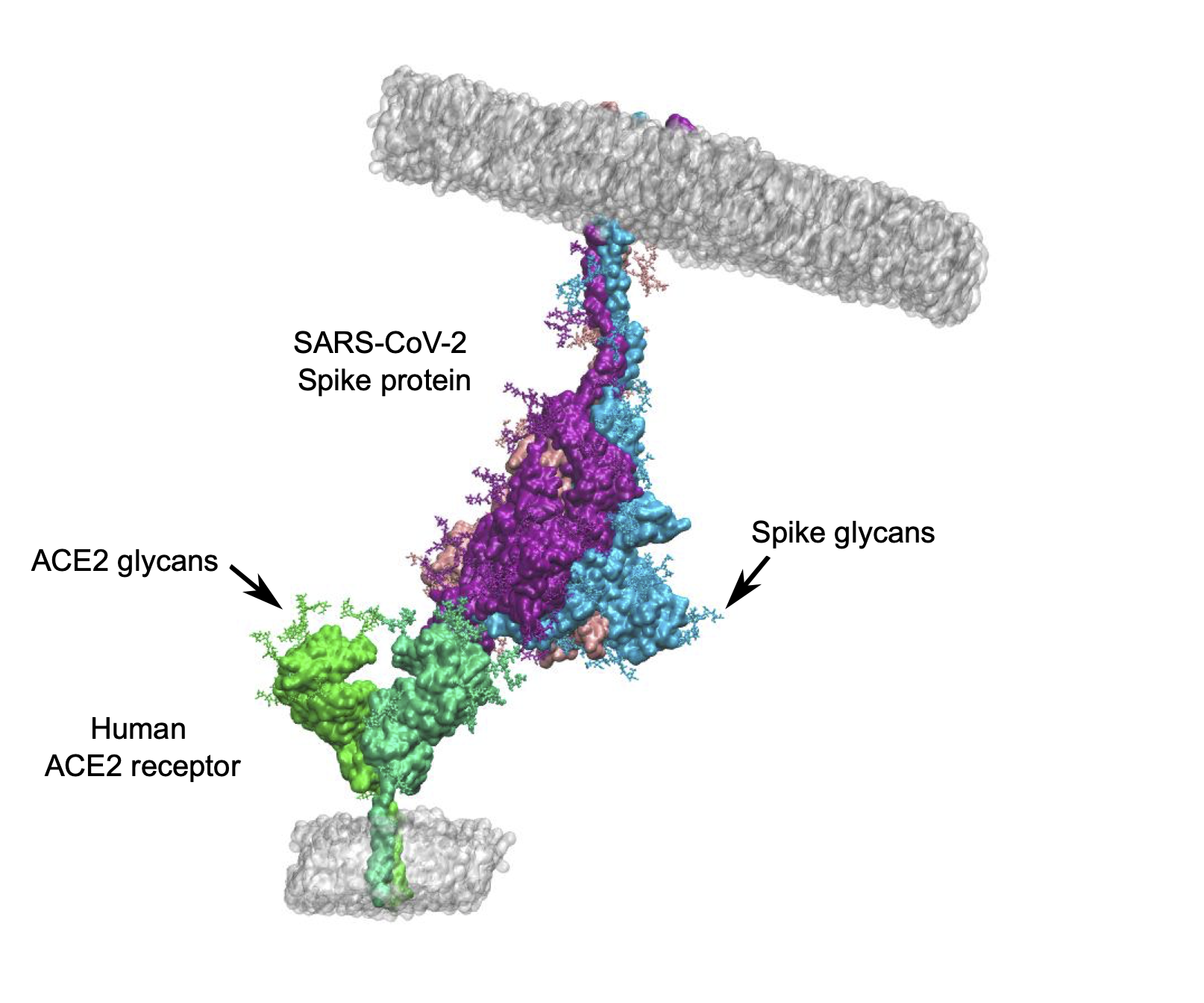
Figure 1: Interaction of the Spike protein of SARS-CoV-2 virus with its human receptor ACE2. Glycans are shown in both proteins.
Conventional MD simulation can help us to study the nature and dynamics of protein-glycan interactions (Figure 2); however, High flexibility of glycan and their week interaction mean that we usually need long timescales (μs to ms) to sample all relevant configurations of the glycan-protein system. These timescales are normally out of reach for conventional equilibrium simulations. Therefore, advanced simulation methods are required that will help us to enhance the sampling along predefined degrees of freedom, denoted as collective variables (CVs). At the end, methods developed based on various concepts in statistical physics can be used to reconstruct the free energy profile as a function of these CVs [4].
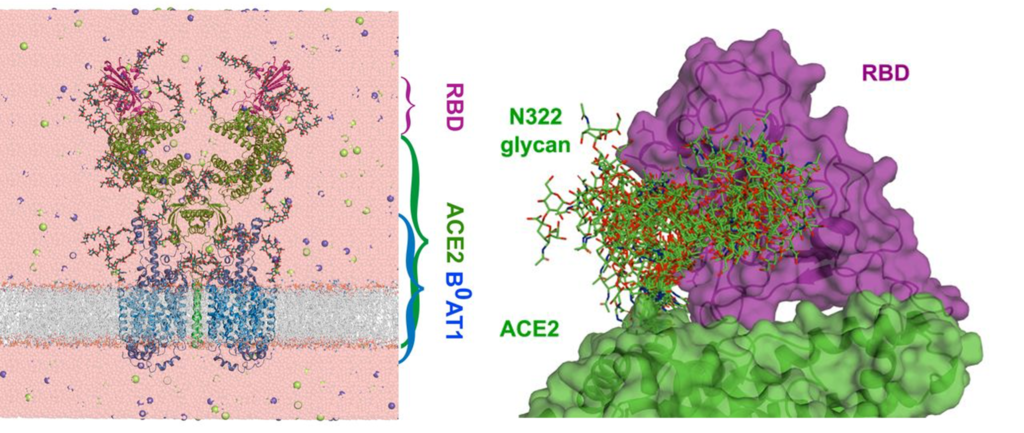
Figure 2: Molecular simulations can reveal the interaction of glycans with the partner protein (ACE2 receptor in binding to SARS-CoV-2 spike receptor binding domain (RBD)) (taken from Ref. 3).
Goal
This proposal aims to understand the glycan contribution to the binding affinity of two interacting protein complexes. We will use a combination of computational approaches, including molecular dynamics simulations and enhanced sampling methods, to calculate accurately the free energy of binding of glycan into a partner protein based on the techniques from statistical mechanics and thermodynamics. The successful estimation of the glycan contribution to the binding affinity will shed light on the pivotal role of glycan in biological processes such as host-cell interactions.
To achieve this goal, this thesis aims to apply various types of molecular dynamics simulations will be performed. These simulations will include both basic molecular dynamics simulations and enhanced molecular dynamics simulations, such as umbrella sampling and metadynamics.
First, we use the structural models of glycoprotein proteins of interest (ACE2 and lysosomal receptors) and their previously run simulations to analyze the glycan-protein interaction. For each of these simulations, the student will analyze the contact network between the glycan and protein and then will calculate the estimated interaction energy will them. Later, the search for optimal parameters to use as collective variables will be performed. The focus of the project is to calculate free energy of binding for glycan using the identified collective variable and enhanced sampling methods (. This will allow us to have a comprehensive understanding of how the presence of glycans affects the binding affinity of the proteins to their interacting partner and use this information for design of effective therapeutic glycoproteins.
The student will analyze the existing MD data using MDAnalysis (MDAnalysis is a python-based package for analysis of MD simulation data). The next step MD simulations, mainly the enhanced sampling, will be performed in an open-source MD-engine GROMACS and using PLUMED (PLUMED is an open-source library that provides a wide range of enhanced-sampling algorithms and free-energy methods). Finally, the student will construct the free energy profile based on methodologies from statistical physics/thermodynamics using PLUMED and ThermoLIB packages (ThermoLIB is a Python-based library to construct free energy surfaces (FES) as a function of CVs from the output of molecular simulations).
- Study programmeMaster of Science in Biomedical Engineering [EMBIEN]Keywordsprotein complexes, glycan binding, Molecular dynamics simulations, Free energy calculations, Statistical physicsReferences
[1] Biomolecules 2015, 5, 2056-2072.
[2] Cell Host Microbe. 2020, 28, 586–601.
[3] PNAS. 2021, 118, e2100425118.
[4] BBA. 2015, 1850, 872-877.
Contact
Ahmad Reza Mehdipour