Quantitative predictions of density and viscosity through force matching
Quantitative predictions of density and viscosity through force matching
Promotor(en): T. Verstraelen, D. Fauconnier /28306 / Model and software developmentBackground and problem
Molecular Dynamics (MD) simulations bridge the gap between microscopic interactions in a condensed phase and its macroscopic properties. A classic example is the Lennard-Jones fluid: using only a simple Lennard-Jones potential for pairs of noble gas atoms, MD predicts properties such as density, viscosity, heat capacity, … as a function of the applied temperature and pressure. Also, the entire phase diagram of a noble gas can be derived in a straightforward manner.
The applicability of MD goes far beyond simple noble gases. For example, also the properties of more complex systems, such as lubricants, can in principle be simulated in a similar fashion. Such simulations are highly relevant because lubricants play a key role in the transition to renewable energy sources, e.g., they are critical for the efficiency of energy conversion in windmills and the lifetime of mechanical parts in their gearboxes. [1,2] A particular challenge for such technologies is to understand lubricant properties at extreme conditions (high temperatures, pressures, and shear rates). Experimental measurements are extremely difficult and expensive, such that MD simulations become an attractive alternative.
The reliability of an MD simulation is completely determined by the accuracy of the forces acting on the nuclei, with which Newton’s equations of motion are integrated. For noble gases, the Lennard-Jones potential is satisfactory for a wide range of conditions, but in fact it is far from perfect. For example, it completely overestimates forces and energies when two noble gas atoms are pushed together. Nevertheless, these errors have a negligible effect on simulation outcomes. For lubricants, comparable (slightly more complicated) models are used in MD simulations, yet the impact of comparable approximations on predicted properties is not well understood.
Goal
For specific configurations of (pairs of) lubricant molecules, it is possible to compute all governing forces accurately with electronic structure methods, yet such calculations are too time-consuming to run MD simulations. For MD, the forces must be computed with simple approximate models, so-called force fields, of which the Lennard-Jones model is a basic example. Naively, one may try to adjust parameters in a force-field model with force matching [3] for a selected set of atom positions:
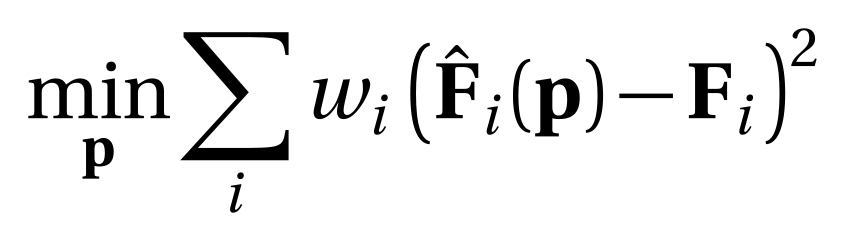
where p is a vector of model parameters. When used blindly, this type of model training will not result in a usable force field. The exact choice of molecular geometries and the weights used to balance the importance of each force have a significant impact on the outcome. It is not clear how to make such choices a priori. The goal of this thesis is to address this difficulty.
In the context of machine learning, one faces a similar issue, yet it is usually hidden by the fact that neural networks are not limited in their number of adjustable parameters. More data and a more complex network will eventually be able to reproduce any data. The same strategy is also applied in machine learning force fields, yet it results in computationally expensive models. One may rephrase the goal of this thesis as follows: which errors are unimportant and can be ignored when training a model? This will be tested initially for the density and viscosity for Argon over a range of temperatures and pressure. For this system, the Lennard-Jones potential and all its properties are well understood, and highly accurate reference data are available. In a second stage, the results for the Argon model will be generalized towards a lubricant model.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Electromechanical Engineering [EMELME], Master of Science in Physics and Astronomy [CMFYST]KeywordsMolecular dynamics, model training, viscosity, densityReferences
[1] Ewen, J. P., Spikes, H. A., & Dini, D. (2021). Contributions of Molecular Dynamics Simulations to Elastohydrodynamic Lubrication. Tribology Letters, 69, 24. 10.1007/s11249-021-01399-w
[2] Meng, Y., Xu, J., Jin, Z., Prakash, B., & Hu, Y. (2020). A review of recent advances in tribology. In Friction (Vol. 8, Issue 2). 10.1007/s40544-020-0367-2
[3] Ercolessi, F. and Adams J. B. (1994). Interatomic Potentials from First-Principles Calculations: The Force-Matching Method, Europhysics Letters, 26, 583. 10.1209/0295-5075/26/8/005
Contact
Toon Verstraelen