Mutation of the electronic quantum genome by approximation effects
Mutation of the electronic quantum genome by approximation effects
Promotor(en): S. Cottenier /15_MAT07 / Solid-state physicsWe are uniquely defined by our genome. Depending on the presence of different genes, it determines how we look and how we act. In the same way, materials are characterized by their electronic structure. The position of the electronic energy bands provide a unique identification of a crystalline material, and as these bands change, so do the properties of the material. A recent article describes how this electronic genome can be translated into an interpretable sequence [1]. This can either be done in a symmetry-dependent way, by discretizing the material's band structure at several high-symmetry point in reciprocal space, or in a way independent of the crystal symmetry, by immediately discretizing the electronic density of states. The resulting sequence makes it possible to compare very different materials, and a close agreement in the electronic genome is expected to lead to very similar properties of another nature as well (e.g. similar mechanical properties, similar magnetic properties, ...).
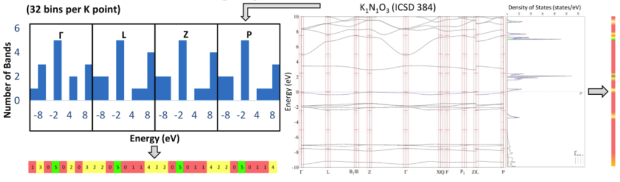
If we now go back to the human genome, we see that every baby is born with a "pure" genetic make-up. Exposure to the environment can cause the genes to change, however. This is why even identical twins are not identical at all, especially as they grow older. The same can be said about the electronic genome of materials. Solving the Schrödinger equation should yield the "pure" electronic band structure, but in practice, the Schrödinger equation cannot be solved exactly (intrinsic errors). Several approximations are available, which not only vary in accuracy, but also in computational load: the more accurately you want to retrieve the pure electronic genome, the higher the cost. Moreover, even within a particular approximation, implementing the governing equations into a computer code requires additional assumptions (numerical errors). Each of these effects can be considered as an environment that influences the pure genome, mutating the individual genes, and hence also the predicted properties of the material.
The Center for Molecular Modeling has gained an international reputation in the quantification of intrinsic and numerical errors [2], and now plays a leading role in a global effort to compare the effect of different approximations within density-functional theory (DFT), a popular approach to solving the Schrödinger equation. Current investigations focus on the influence on the energy-volume relation of different materials [3], but this does not capture the full effect on the electronic band structure. By assessing intrinsic and numerical errors based on the electronic genome, you may not only discover essential changes, but also identify the crucial genes that determine a material's behaviour.
Goal In this thesis, you will apply DFT in different approximations to investigate the influence on the electronic genome. You will initially look at purely elemental crystals to see how using all-electron methods (the best available approximation of zero numerical errors) compares to potential-based methods, which employ a smoothing potential for the electronic wave function near the nucleus. These potentials are often tuned to a few energy levels, so this should be visible from the sequenced electronic genome. Moreover, to compare the different genomes quantitatively, you will need to find an appropriate similarity measure. Finally, you can also investigate how genome changes affect other materials properties, such as mechanical resistance or crystal volume.
In a second stage, you can have a look at how the electronic genome is transferred to the offspring. Indeed, just like parts of the genome of both parents are used to build the genome of the child, the electronic genome of the elemental crystals A en B will be linked to that of multicomponent compound AB. It is up to you to discover how similar the child material is to the parents, and whether the sensitivity of particular genes to numerical errors is inherited too.
Finally, it can also be interesting to investigate the effect of different exchange-correlation functionals (or functionals in short). The functional is the source of intrinsic errors in DFT, and its effect on the electronic genome is expected to be of an entirely different nature than that of numerical errors. Nevertheless, if systematic gene mutations could be discovered, this would be a great help for developers of functionals, ideally leading to methods with a minimum deviation from the pure genome.
Physics & Engineering aspects:
Physics aspect: use of quantum physical methods to characterize materials
Engineering aspect: application to the properties of metals and semiconductors

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) Modelling, materials, nanoReferences
[1] O. Isayev et al., 'Materials Cartography: Representing and Mining Materials Space Using Structural and Electronic Fingerprints', Chem. Mater. 27, 735-743 (2015). http://dx.doi.org/10.1021/cm503507h
[2] K. Lejaeghere et al., 'Error Estimates for Solid-State Density-Functional Theory Predictions: An Overview by Means of the Ground-State Elemental Crystals', Crit. Rev. Solid State 39, 1-24 (2014). http://dx.doi.org/10.1080/10408436.2013.772503
[3] https://molmod.ugent.be/deltacodesdft
Contact
Stefaan Cottenier