Molecular-level understanding of phase transformations and free energy profiles of flexible materials
Molecular-level understanding of phase transformations and free energy profiles of flexible materials
Promotor(en): V. Van Speybroeck, A. Ghysels /15_NANO03 / Nanoporous materialsNanoporous materials are omnipresent in a broad versatility of application domains such as energy storage, energy conversions, sustainable chemistry, biomedical applications,… Engineering these materials towards the desired applications is of utmost importance. Molecular modeling plays a pivotal role in this process, since it can unravel the molecular factors that control the observed macroscopic properties. One emerging class of materials are metal-organic frameworks (MOFs). The discovery of metal-organic frameworks (MOFs) was a milestone in material science and computational physics. These hybrid materials consist of metal-oxide clusters interconnected by means of organic linkers, resulting in a 3D periodic framework with nanosized pores (nanoporous crystal). Due to the porosity, these materials show many promising applications such as capture, separation and detection of gasses.
A property which is very intriguing is the breathing capacity of these materials, i.e. their ability to undergo large structural deformations upon external stimuli. (Figure 1) This special type of behavior is unique and is known in literature as framework breathing. This property opens a new range of applications such as nano dampeners and nano springs. The highly flexible nanoporous MIL-53 type of materials (MIL for Material of Institut Lavoisier) has received considerable attention within this respect. It is however very difficult to get track of the molecular factors controlling the phase transitions. Within the Center for Molecular Modeling, various force field models have been developed that enable the description of the energy variations observed during phase transitions. However the true challenge relies in the estimation of the free energy of the materials at realistic conditions of temperature, pressure and realistic loadings of guest molecules. In the proposed program the student will use and develop advanced modeling techniques to compute the free energies that control the phase behavior of flexible metal organic frameworks.
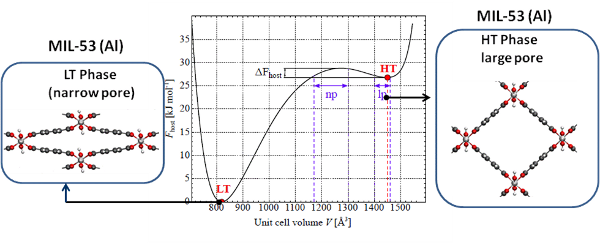
Recently, a thermodynamic model has been proposed at the CMM to describe the breathing behavior of metal-organic frameworks under influence of adsorption of guest molecules. This model is able to predict the framework transformations (breathing) stimulated by external mechanical pressure or adsorption of guest molecules. However, a missing factor is the temperature-induced breathing. A crucial part of the model is the free energy profile of the empty MOF framework. Up to now, these free energy profiles are computed at fixed temperature and the influence of the temperature is not clear. In this thesis, the student will first investigate this temperature dependence by computing the free energy (and hence entropy) at various temperatures. Next, the student will try to capture the temperature dependence by proposing an analytical temperature-dependent variant of the thermodynamical model. The temperature dependence of the entropy is of particular interest due to its close relation with the heat capacity and therefore the nuclear quantum effects (see below).
The calculation of the free energy profiles will be performed using advanced molecular dynamics simulations that enable to sample phase transitions of these flexible materials. Special techniques such metadynamics, thermodynamic integration,… will have to be used to sample also the transitions between various phases. One point of interest concerns the molecular dynamics simulations itself, in which the nuclei are treated as classical particles moving along a potential energy surface. The dynamics results from the Newton’s equation of motions. However some MOFs have some high lying vibrational modes for which it is yet unknown whether these motions have a profound effect on the free energy and entropy. The impact of these nuclear quantum effects will be investigated on the nature of the free energy profile by means of dedicated a theoretical framework that combines coloured-noise MD techniques with the path integral formalism.
As a result of the advanced molecular dynamics simulations, a thorough understanding of the free energy of flexible materials will be obtained in terms of temperature. As a result, it will be possible to assess for the first time the influence of the entropy on the observed phase transition in these material.
The research will be performed in the framework of a larger concerted action to simulate transformations in nanoporous materials. The Center for Molecular Modeling has built up vast expertise in these advanced simulation techniques and collaborates on the subject with leading experimental and theoretical partners. It is the intention to involve the student actively in the work discussions with our collaborators. The CMM has access to sufficient computational resources to execute this research project. The proposed topic is challenging and requires profound theoretical physical insights, technical skills, creativity.
This topic is on the cross road between statistical physics, thermodynamics and molecular simulation. The topic will deepen your knowledge about simulation techniques and computational physics. If needed, the required programming skills will be transfered during the thesis.
Engineering & Physics aspects:
Physics: use of quantum and classical mechanical models for materials modelling
Engineering: engineering of materials for applications such as storage, separation, …

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) modeling, materials, nanoKeywordsMolecular simulation, Free energy calculations, Nanoporous materials
Contact
Louis Vanduyfhuys
An Ghysels
Veronique Van Speybroeck